Metabolisme
Når vi får i oss et legemiddel, starter en reise gjennom kroppen hvor stoffet skal absorberes, transporteres, virke – og til slutt fjernes. Denne siste delen av reisen er det kroppen selv som styrer gjennom prosesser vi kaller metabolisme.
Målet er enkelt: å gjøre stoffet mer vannløselig, slik at det kan skilles ut via urin eller galle.
Men metabolisme handler ikke bare om å rydde opp. I noen tilfeller aktiverer kroppen et legemiddel gjennom metabolismen – og i andre tilfeller kan det skapes giftige metabolitter. Å forstå hvordan og hvor dette skjer, er avgjørende både for trygg legemiddelbruk og for utvikling av nye medisiner.
Hva er metabolisme?
Metabolisme, eller mer presist biotransformasjon, er en enzymatisk prosess der kroppen omdanner kjemiske stoffer. Disse stoffene kan være kroppens egne (som hormoner), eller de kan være fremmede for kroppen – det vi kaller xenobiotika. Dette inkluderer legemidler, miljøgifter, kosttilskudd og kunstige søtstoffer.
Biotransformasjonen foregår hovedsakelig i leveren, og endrer kjemiske egenskaper hos stoffene. Målet er som regel å gjøre dem mindre biologisk aktive og mer vannløselige, slik at de lettere kan skilles ut.
Prosessen kan ha flere ulike utfall:
- Noen stoffer inaktiveres fullstendig.
- Andre omdannes til aktive metabolitter – dette er ofte tilfellet for såkalte prodrugs, legemidler som først aktiveres i kroppen.
- I enkelte tilfeller dannes toksiske metabolitter, som kan føre til bivirkninger eller organskade.
- Til slutt kan det dannes konjugater, som er vannløselige forbindelser bundet til kroppens egne stoffer, klare for utskillelse.
Hvor i kroppen skjer metabolismen?
Selv om leveren med god grunn får hovedrollen når vi snakker om legemiddelmetabolisme, er den langt fra alene om oppgaven. Kroppen vår har et nettverk av enzymer fordelt på ulike vev og organer, alle med sin rolle i å omdanne stoffer – enten for å aktivere dem, avgifte dem, eller gjøre dem klare til utskillelse. Disse enzymene kan deles inn etter hvor de befinner seg, og hvilken type reaksjoner de katalyserer.
Leveren er det viktigste organet for legemiddelmetabolisme, og det er her mesteparten av omdanningen foregår.
Levercellene (hepatocyttene) inneholder et bredt spekter av enzymer som utfører både fase I- og fase II-reaksjoner. Disse enzymene finnes i ulike strukturer inne i cellen, og kan grovt deles i to hovedgrupper: mikrosomale og ikke-mikrosomale enzymer.
Mikrosomale enzymer i leveren
Blant de mikrosomale enzymene finner vi den mest kjente og viktigste enzymfamilien i legemiddelmetabolismen: cytokrom P450 (CYP). Disse enzymene befinner seg i det glatte endoplasmatiske retikulum, og står for størstedelen av oksidasjonsreaksjonene i fase I. CYP-enzymer er spesialister i å endre strukturen til molekyler ved å oksidere dem, og de kan både aktivere, inaktivere og i noen tilfeller gjøre stoffer mer toksiske.
Ikke-mikrosomale enzymer i leveren
I tillegg til CYP-systemet har leveren også en rekke ikke-mikrosomale enzymer, som befinner seg i cytoplasma og mitokondrier. Disse enzymene står for andre typer reaksjoner, blant annet:
- Acetylering, som innebærer overføring av en acetylgruppe
- Sulfatering, der en sulfatgruppe kobles på molekylet
- Hydrolyse, hvor molekyler spaltes ved hjelp av vann
- Dehydrogenering, særlig av alkohol og aldehyder
Disse reaksjonene er viktige, særlig for stoffer som ikke metaboliseres av CYP-systemet, og utgjør en betydelig del av kroppens kapasitet for avgiftning.
De to fasene i legemiddelmetabolismen
Metabolismen av legemidler og fremmedstoffer i kroppen foregår som regel i to trinn: fase I og fase II. Ikke alle stoffer må gjennom begge fasene, men oftest skjer de i rekkefølge. Fase I-reaksjonene forbereder stoffet for videre behandling, mens fase II fullfører prosessen og gjør det klart for utskillelse. Sammen sørger disse fasene for at kroppen kan håndtere og kvitte seg med både nyttige og potensielt skadelige forbindelser.
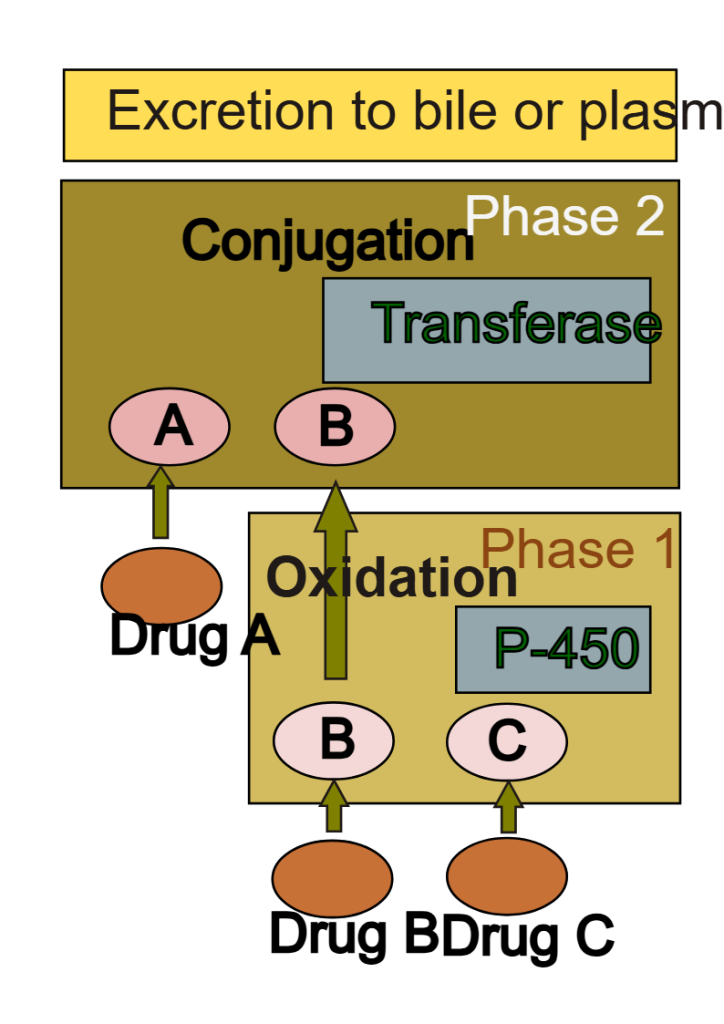
Fase I: Funksjonell modifisering
I den første fasen av metabolismen skjer det en kjemisk modifisering av molekylet. Dette innebærer enten at kroppen legger til nye grupper på molekylet, eller at den avdekker funksjonelle grupper som allerede finnes, men som har vært «skjult» i strukturen. De vanligste funksjonelle gruppene som blir introdusert eller eksponert er:
- Hydroksylgrupper (-OH), sulfhydrylgrupper (-SH), aminogrupper (-NH₂) og karboksylgrupper (-COOH)
Disse gruppene gjør molekylet mer vannløselig og klargjør det for videre behandling i fase II.
Reaksjonene som skjer i fase I, kan deles inn i tre hovedtyper:
- Oksidasjon – den klart vanligste reaksjonen, hvor molekylet mister elektroner eller får lagt til oksygen
- Reduksjon – det motsatte av oksidasjon; molekylet tar opp elektroner
- Hydrolyse – molekylet spaltes ved hjelp av vann
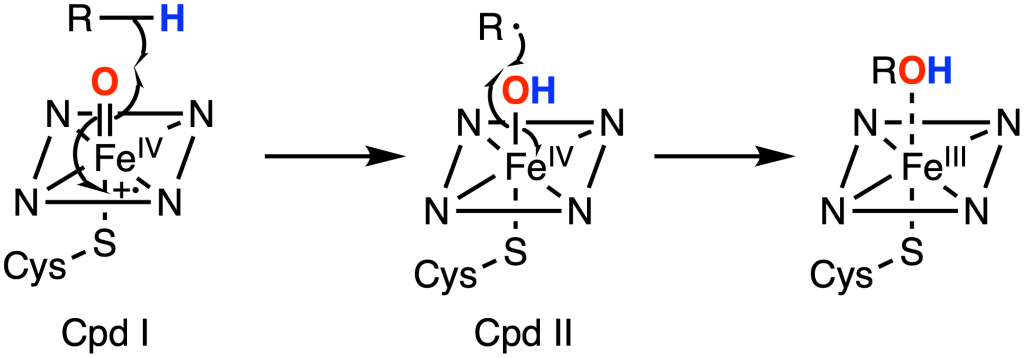
Et viktig trekk ved fase I er at det er her CYP-enzymer (cytokrom P450) spiller sin hovedrolle. Disse enzymene utgjør den viktigste familien av oksiderende enzymer i leveren, og står for metabolismen av rundt 75 % av alle legemidler som brukes i klinisk praksis.
CYP-enzymene befinner seg hovedsakelig i det glatte endoplasmatiske retikulum i hepatocytter, og deres hovedoppgave er å gjøre fettløselige molekyler mer vannløselige ved å innføre oksygenatomer i strukturen.
De bruker oksygen (O₂) og elektroner for å omdanne legemidler, miljøgifter og kroppens egne stoffer.
Et enkelt legemiddel kan bli metabolisert av flere CYP-enzymer. Et godt eksempel er acetaminophen, som omdannes av en rekke ulike CYP-er: CYP1A2, CYP2E1, CYP2D6 og CYP3A4. Hvilken vei stoffet tar, kan påvirkes av genetikk, legemiddelinteraksjoner og til og med kosthold.
Andre eksempler på stoffer som metaboliseres av flere CYP-enzymer:
- Koffein: av CYP1A2, CYP2E1, CYP3A4
- Warfarin: av CYP1A1, CYP1A2, CYP2C9, CYP3A4
- Testosteron: av CYP1A1, CYP2C9, CYP3A4, CYP2A6
Dette har viktige kliniske konsekvenser. Hvis et annet legemiddel hemmer ett av enzymene, kan metabolismen forsinkes og føre til økt effekt eller toksisitet. Dette kalles legemiddelinteraksjon, og CYP-systemet er ofte involvert.
Utfallet av fase I varierer:
- Stoffet kan bli inaktivt, slik at det mister sin farmakologiske virkning
- Det kan bli aktivt, slik som ved omdanningen av kodein til morfin
- Eller det kan bli toksisk, som i tilfellet med acetaminophen (paracetamol) som omdannes til den skadelige metabolitten NAPQI
I noen tilfeller er molekylet etter fase I allerede tilstrekkelig vannløselig og klart for utskillelse. I de fleste tilfeller går det derimot videre til neste fase.
Fase II: Konjugering
Når et molekyl har gjennomgått funksjonell modifisering i fase I, er det ofte fortsatt for fettløselig til å kunne skilles ut effektivt.
Da trår fase II-metabolismen til. I denne fasen kobler kroppen det bearbeidede molekylet sammen med et nytt, vannløselig molekyl.
Dette kalles konjugering, og er ofte kroppens siste steg før stoffet sendes ut med urin eller galle.
Konjugeringen skjer gjennom spesifikke enzymreaksjoner, og avhengig av hvilket enzym og hvilket substrat som brukes, finnes det flere ulike konjugeringsveier.
Konjugering skjer med kroppens egne stoffer, ofte små molekyler som lett kan kobles til metabolitten. De vanligste konjugeringspartnerne er:
- Glukuronsyre (glucuronid), sulfat, acetat, aminosyrer (for eksempel glycin eller taurin) og glutathion (GSH) – spesielt viktig for å avgifte toksiske metabolitter
Konjugeringen utføres av spesialiserte enzymer, og en viktig gruppe her er glutathion-S-transferaser (GST). Disse enzymene beskytter kroppen mot skade ved å nøytralisere reaktive forbindelser, særlig ved overdoser eller ved høy eksponering for miljøgifter.
Etter konjugering er stoffet som regel biologisk inaktivt, og i tillegg svært vannløselig. Dermed kan det trygt skilles ut, oftest via nyrene i urinen, men også gjennom galle og avføring.
De viktigste konjugeringsreaksjonene
Det finnes mange reaksjoner som:
Sulfatering, acetylering, aminosyrekonjugering, glutation-konjugering, fettsyrekonjugering og kondenseringsreaksjoner.
Men blant alle konjugeringsreaksjonene er det glukuronidering som er den mest dominerende. Dette er kroppens hovedvei for å gjøre mange legemidler, hormoner og giftstoffer vannløselige.
Glukuronidering skjer i leveren, og enzymet som utfører reaksjonen – UDP-glukuronosyltransferase (UGT) – finnes i mange varianter med ulik substratspesifisitet.
Når kroppen står overfor toksiske metabolitter, som for eksempel NAPQI, trer enzymet glutathion-S-transferase (GST) i kraft.
Det kobler glutation til det reaktive molekylet og gjør det vannløselig og ufarlig.
Hvis glutationlagrene blir tomme (for eksempel ved overdose av paracetamol), kan de toksiske metabolittene skade celler – særlig leverceller. Reaksjonen er energikrevende, men svært effektiv. Resultatet er et glukuronid-konjugat som skilles raskt ut via nyrene.
Metabolsk aktivering – når metabolisme skaper gift
Vi tenker ofte på metabolisme som en måte kroppen avgifter fremmedstoffer på. Men i enkelte tilfeller gjør metabolismen det motsatte: den aktiverer et ellers ufarlig stoff til en reaktiv og farlig metabolitt. Dette kalles metabolsk aktivering eller bioaktivering.
Et klassisk eksempel er kjemikalier i sigarettrøyk, som benzo[a]pyren.
Dette stoffet i seg selv er relativt lite reaktivt, men etter oksidasjon av CYP-enzymer dannes et epoksid som kan binde seg til DNA og forårsake mutasjoner – en mulig vei til kreft.
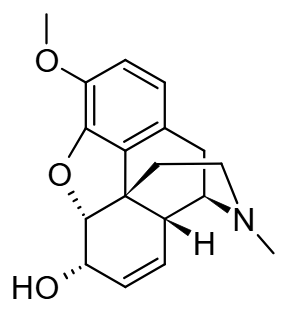
Det finnes også mange prodrugs – farmakologisk inaktive legemidler som må aktiveres av kroppens metabolisme for å virke.
Et prodrug er et legemiddel som gis i inaktiv form, men som gjennom enzymatisk metabolisme omdannes til et aktivt legemiddel. Dette brukes for å:
- Øke biotilgjengelighet
- Redusere bivirkninger i mage-tarm
- Sikre at legemidlet aktiveres først der det trengs
Eksempler:
- Kodein omdannes til morfin via CYP2D6
- Levodopa omdannes til dopamin i hjernen (dopamin i seg selv kan ikke krysse blod–hjerne-barrieren)
Aktiveringen skjer ofte gjennom hydrolyse av en ester- eller amidbinding, men kan også skje via oksidasjon eller reduksjon.
Faktorer som påvirker legemiddelmetabolismen
Selv om enzymene som driver legemiddelmetabolismen i utgangspunktet er genetisk programmert, er de langt fra stabile og forutsigbare. Tvert imot – aktiviteten til disse enzymene varierer betydelig fra person til person, og fra situasjon til situasjon. Dette skaper store individuelle forskjeller i hvordan legemidler tas opp, virker og elimineres.
En av de viktigste ytre faktorene som påvirker metabolismen, er eksponering for andre stoffer i miljøet. Dette inkluderer:
- Legemidler og rusmidler
- Mat, urter og kosttilskudd
- Miljøgifter, som pesticider eller industrielle kjemikalier
Disse stoffene kan påvirke enzymaktiviteten på to måter:
Induksjon av enzymer
Noen xenobiotika (fremmedstoffer) kan påvirke genuttrykket til enzymene våre. Dette skjer ved at stoffene binder seg til reseptorer som aktiverer transkripsjonsfaktorer, som igjen øker transkripsjonen av CYP-gener.
Resultatet er økt mengde enzym, som fører til raskere metabolisme av legemidler.
Eksempler på stoffer som induserer CYP-enzymer:
- Fenobarbital (et antiepileptikum), Johannesurt (et naturlig antidepressivum) og sigarettrøyk (induserer blant annet CYP1A2).
Dette kan ha flere viktige konsekvenser:
- Legemidler brytes ned raskere enn vanlig
- Plasmakonsentrasjonen reduseres
- Førstepassasje-effekten øker, noe som fører til lavere mengde aktivt stoff som når systemisk sirkulasjon
- Biotilgjengeligheten synker, og effekten av legemidlet kan reduseres
Hvis metabolitten som dannes er inaktiv, vil virkningen av legemidlet bli svekket. Dersom metabolitten derimot er aktiv eller toksisk, kan raskere metabolisme føre til uønskede bivirkninger eller forgiftning.
Inhibering – når kroppen bremser ned
I kontrast til induksjon finner vi enzymhemming, eller inhibering. Her skjer det en reduksjon i enzymaktiviteten, enten ved at et stoff konkurrerer med et annet om å binde seg til enzymet (kompetitiv hemming), eller ved at enzymet inaktiveres mer varig (irreversibel hemming).
Når metabolismen hemmes, brytes legemidler ned langsommere. Dette fører ofte til:
- Økt plasmakonsentrasjon av modersubstansen
- Redusert produksjon av metabolitter
- Økt og forlenget farmakologisk effekt
- Økt risiko for toksiske bivirkninger
Et velkjent og klinisk relevant eksempel er grapefruktjuice. Juicen inneholder furanokumariner og flavonoider som hemmer CYP3A4, spesielt i tarmveggen. Dette fører til redusert førstepassasje-metabolisme av mange legemidler, og gir økt plasmakonsentrasjon. I praksis kan dette doble effekten av enkelte legemidler, og i verste fall føre til overdosering – uten at dosen er endret.
En lignende effekt er vist med tranebærjuice, som kan hemme CYP-enzymer og øke plasmanivåene av legemidler som warfarin.
Kjønn, alder og genetikk
Selv om legemidler forskrives i standardiserte doser, er det ingen hemmelighet at virkningen kan variere dramatisk fra person til person. Noen opplever kraftig effekt og bivirkninger, mens andre merker knapt noe i det hele tatt – selv med samme dose. En viktig del av forklaringen ligger i biologiske forskjeller, og særlig tre faktorer skiller seg ut: kjønn, alder og genetiske variasjoner.
Menn og kvinner har forskjellig hormonprofil, kroppssammensetning og genuttrykk – og dette påvirker også legemiddelmetabolismen. Spesielt kjønnshormonene, som østrogen og testosteron, ser ut til å modulere uttrykket av visse CYP-enzymer.
Flere studier har vist at kvinner i gjennomsnitt har lavere aktivitet av enkelte CYP-enzymer sammenlignet med menn. Dette betyr at kvinner i noen tilfeller vil bryte ned legemidler langsommere, noe som kan føre til høyere plasmakonsentrasjoner og økt risiko for bivirkninger – eller, omvendt, til svakere effekt hvis aktiv metabolitt ikke produseres effektivt.
Samtidig varierer hormonprofilen gjennom livet, som ved menstruasjonssyklus, graviditet og overgangsalder, noe som gjør kvinners metabolisme mer dynamisk over tid. Dette er viktig å ta hensyn til både i forskning og klinikk, men blir ofte oversett i standard behandlingsregimer.
Alder
Alder er en annen sentral faktor for legemiddelmetabolisme, og påvirkningen er mest uttalt i begge ender av aldersspekteret: hos nyfødte og hos eldre.
Hos nyfødte er leveren fortsatt umoden, og mange av enzymene som normalt deltar i fase I- og fase II-metabolismen, er enten lite uttrykt eller fungerer dårlig. Det betyr at nyfødte har en nedsatt evne til å metabolisere og skille ut legemidler, og derfor er de svært følsomme for både dose og legemiddelvalg. Dette er grunnen til at man ofte bruker spesialtilpassede doser hos spedbarn, og alltid med forsiktighet.
Hos eldre mennesker skjer det en gradvis reduksjon i leverens masse og blodgjennomstrømning, som igjen reduserer leverens totale metabolisme-kapasitet. Dette påvirker særlig legemidler med høy lever-clearance og høy førstepassasjeeffekt. Kombinert med aldersrelatert nyresvikt, kan dette føre til opphopning av aktive stoffer eller toksiske metabolitter. Derfor bør mange legemidler doseres lavere eller overvåkes tettere hos eldre pasienter.
Genetikk og polymorfismer – individets unike enzymprofil
Den kanskje mest fascinerende og avgjørende faktoren for individuell variasjon i legemiddelmetabolisme er genetikken. Mange av de viktigste CYP-enzymene finnes i flere genetiske varianter, kjent som polymorfismer. Dette innebærer små forskjeller i DNA-sekvensen som kan føre til store forskjeller i enzymaktivitet.
Genetiske variasjoner kan gi:
- Ultrarask metabolisme: Noen individer har flere kopier av genet som koder for et gitt enzym, og bryter dermed ned legemidler ekstremt raskt. Resultatet er ofte redusert effekt, fordi legemidlet ikke får virke lenge nok i kroppen.
- Langsom metabolisme: Andre har mutasjoner som gjør enzymet mindre effektivt, eller tregere i funksjonen. Dette fører til høyere plasmakonsentrasjoner og økt risiko for bivirkninger eller toksisitet.
- Manglende metabolisme: I noen tilfeller fungerer ikke enzymet i det hele tatt. Dette kan gi total svikt i aktivering eller nedbrytning av legemidler.
Eksempel: CYP2D6 og kodein
Et av de best kjente eksemplene på genetisk variasjon er CYP2D6, et enzym som er avgjørende for aktivering av kodein til morfin. CYP2D6 har over 100 kjente genetiske varianter, og basert på disse deles befolkningen inn i ulike metaboliseringskategorier:
- Ultraraske metaboliserere omdanner kodein til morfin så raskt at de risikerer kraftige bivirkninger – og i noen tilfeller toksiske nivåer.
- Langsomme metaboliserere klarer nesten ikke å produsere morfin, og får derfor liten eller ingen smertelindring av kodein.
Dette eksemplet illustrerer hvor viktig det er å kjenne pasientens genetiske profil, spesielt når man bruker legemidler med smalt terapeutisk vindu eller som krever enzymatisk aktivering.
Kliniske eksempler på legemiddelmetabolisme
Teori blir mer håndfast når vi ser på konkrete eksempler. To stoffer som illustrerer ulike sider ved metabolismen, er aspartam, et kunstig søtningsmiddel, og acetaminophen (paracetamol), et av verdens mest brukte smertestillende legemidler. Det første viser hvordan normale metabolske veier kan skape bekymring og misforståelser, mens det andre illustrerer hvordan metabolismen kan gjøre et trygt legemiddel potensielt livstruende.
Aspartam – et søtningsstoff med sterk metabolsk profil
Aspartam er et kunstig søtningsmiddel som brukes i en rekke sukkerfrie produkter, blant annet i brus som Pepsi Max og sukkerfrie tyggegummier. Kjemisk sett er det en dipeptidforbindelse, og når det inntas, spaltes det raskt i tre bestanddeler:
- Fenylalanin
- Asparaginsyre
- Metanol
Alle tre komponentene finnes naturlig i vanlig mat, og kroppen har gode mekanismer for å håndtere dem. Likevel har aspartam fått et ufortjent dårlig rykte, og har i ulike medier blitt beskyldt for å forårsake alt fra hodepine, vektøkning og metabolsk syndrom til kreft og nevrologiske sykdommer. Påstander som «aspartam gir hull i hjernen» eller at det skaper «sur kropp», har florert – ofte uten vitenskapelig belegg.
Flere store metaanalyser og vurderinger fra anerkjente instanser som The European Food Safety Authority (EFSA) og Vitenskapskomiteen for mat (VKM) har slått fast at aspartam er trygt i de mengdene det normalt konsumeres. Inntaksgrensen for aspartam er satt til 40 mg/kg kroppsvekt per dag. For en voksen person tilsvarer dette flere liter sukkerfri brus – langt over det som konsumeres av folk flest.
Et unntak: Føllings sykdom (fenylketonuri)
For pasienter med fenylketonuri (PKU), en medfødt sykdom der kroppen ikke klarer å bryte ned fenylalanin, kan aspartam være farlig. For disse personene må all tilførsel av fenylalanin begrenses, og produkter med aspartam må unngås fullstendig. Derfor merkes slike produkter alltid med en advarsel.
Acetaminophen (Paracetamol) – trygg i små doser, farlig i store
Acetaminophen, kjent som paracetamol i Norge, er et reseptfritt legemiddel brukt mot smerter og feber. Det regnes som trygt og effektivt i anbefalte doser, men er samtidig den vanligste årsaken til legemiddelforgiftninger i Norge.
Statistikk fra klinisk virkelighet:
- Rundt 1500 henvendelser årlig til Giftinformasjonen skyldes overdose med acetaminophen.
- Omtrent 1000 personer legges inn på sykehus hvert år grunnet forgiftning.
- Hvert år registreres det flere dødsfall, til tross for tilgjengelig motgift.
Når acetaminophen inntas i normale doser, skjer metabolismen hovedsakelig i leveren, gjennom konjugeringsreaksjoner:
- Omtrent 60 % konjugeres med glukuronsyre
- Omtrent 35 % konjugeres med sulfat
Disse metabolittene er inaktive og skilles raskt ut gjennom nyrene.
Men en liten andel – typisk 5–10 % – metaboliseres av CYP-enzymer, særlig CYP2E1, CYP1A2 og CYP3A4, til en reaktiv mellomform kalt NAPQI (N-acetyl-p-benzoquinonimin). Under normale forhold nøytraliseres NAPQI raskt ved konjugering med glutation (GSH).
Ved inntak av store doser acetaminophen, eller ved samtidig bruk av stoffer som induserer CYP2E1 (som alkohol eller isoniazid), produseres mer NAPQI enn leveren klarer å nøytralisere. Når glutationlageret tømmes, begynner NAPQI å reagere med cellulære proteiner og lipider, noe som fører til:
- Protein-addukter
- Oksidativt stress
- Celleskade og nekrose
Skaden oppstår særlig i sone 3 i leveren – den delen av leverlobulen som ligger nærmest den sentrale venen. Her er oksygennivået lavt, men CYP-enzymaktiviteten høy. Dette gjør sone 3 ekstra utsatt for NAPQI-indusert skade.
Mikroskopiske bilder av leveren viser tydelig hvordan sone 3 kan nekrotisere ved alvorlig forgiftning, mens sone 1, som ligger nærmest portåren, ofte er intakt.
Behandling: N-acetylcystein
Behandlingen ved acetaminophenforgiftning er rask administrasjon av N-acetylcystein (NAC), som fungerer som en forløper til glutation. Ved å gi NAC kan man gjenopprette glutationnivåene og dermed nøytralisere NAPQI før det gjør skade.
Tiden er avgjørende. NAC må helst gis innen 10 timer etter inntak, og jo tidligere det gis, desto bedre er prognosen.
📚 Anki-kort
Obs, tomt! Kommer etterhvert <3
📝 Eksamensoppgaver
Obs, tomt! Kommer etterhvert <3
👨⚕️ Klinisk case
Obs, tomt! Kommer etterhvert <3
Legg igjen en kommentar