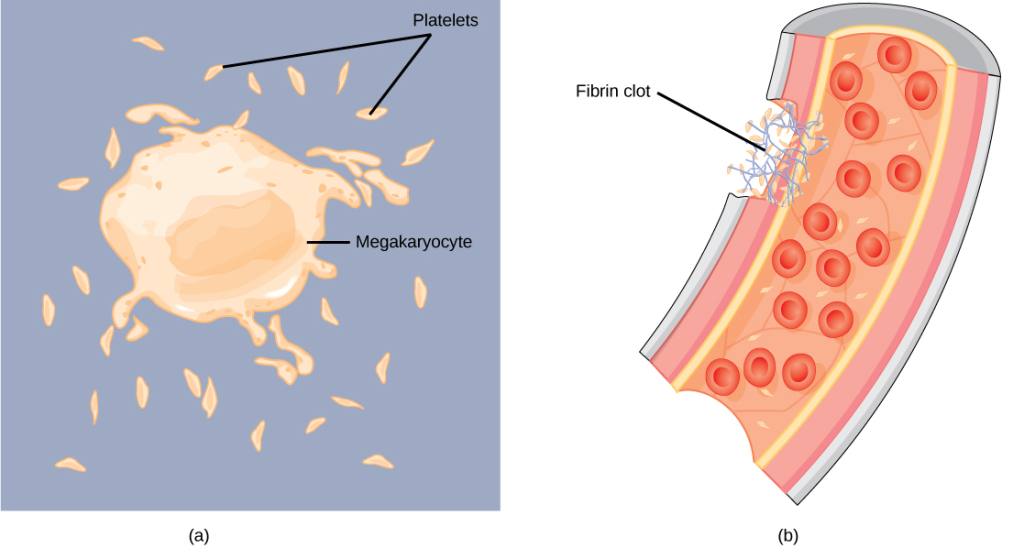
Når kroppen utsettes for en skade som fører til at en blodåre brytes, oppstår en umiddelbar risiko: blod kan lekke ukontrollert ut av sirkulasjonssystemet. Uten et raskt og effektivt mottiltak ville selv små kutt kunne føre til alvorlig blodtap. Løsningen er hemostase – kroppens eget system for å stanse blødning, kontrollert og presist.
Hemostase betyr bokstavelig talt «å stanse blod» – fra gresk hemo (blod) og stasis (stillstand). Dette systemet består av flere mekanismer som sammen sørger for å tette lekkasjen, bevare sirkulasjonen og forhindre at kroppen mister for mye blod. Samtidig må systemet være finjustert: for mye koagulasjon kan føre til blodpropper som igjen blokkerer blodtilførselen til viktige organer. Hemostase er altså en balansekunst mellom å stanse blødning og å bevare normal blodflyt.
Hemostasen deles gjerne inn i to faser, selv om de skjer parallelt:
Den første fasen kalles primær hemostase, og handler om blodplatenes reaksjon på karskaden. I løpet av sekunder fester blodplatene seg til skadestedet, aktiveres og klumper seg sammen i en midlertidig plugg som reduserer blodlekkasjen.
Den andre fasen er sekundær hemostase, og innebærer aktivering av en rekke koagulasjonsfaktorer i blodet. Disse danner et nettverk av fibrintråder som forsterker og stabiliserer den svake platepluggen, slik at blødningen virkelig stanses. Dette fibrinnettet gjør pluggen robust nok til å tåle trykket fra blodstrømmen.
Men prosessen stopper ikke der. Når skaden er reparert, må kroppen også kunne fjerne blodproppen igjen. Dette skjer gjennom fibrinolyse – et enzymatisk system som gradvis bryter ned fibrin og oppløser koagelet slik at blodstrømmen kan gå uhindret igjen. Fibrinolyse sørger for at koagulasjonen ikke varer lenger enn nødvendig.
Hvis noen av disse trinnene svikter, kan det få alvorlige konsekvenser. Manglende hemostase kan gi blødninger som ikke stanser, slik man ser ved sykdommer som hemofili og von Willebrands sykdom. På den andre siden kan for mye aktivering av koagulasjonssystemet føre til blodpropper, som igjen kan gi hjerteinfarkt, lungeemboli eller hjerneslag.
Hemostase er et livsviktig system som må tre i kraft umiddelbart ved skade. Den stopper blødning, beskytter mot blodtap og bevarer sirkulasjonen. Når mekanismene svikter, oppstår enten ukontrollert blødning eller blodpropp, med potensielt livstruende følger. Både i hverdagsfysiologi og under akuttmedisinske situasjoner – som ved kirurgi, traumer og alvorlig sykdom – er en presis regulering av hemostase og fibrinolyse avgjørende.
Primær hemostase
Når en blodåre skades, utsettes det underliggende vevet for blodstrømmen. Det som tidligere var skjult bak et intakt endotel, blir nå eksponert: kollagenfibre, vevsfaktor og andre substanser som normalt ikke kommer i kontakt med sirkulerende blod. Kroppen registrerer dette som en akutt fare, og reagerer umiddelbart med to tiltak: karspasme og aktivering av trombocytter.
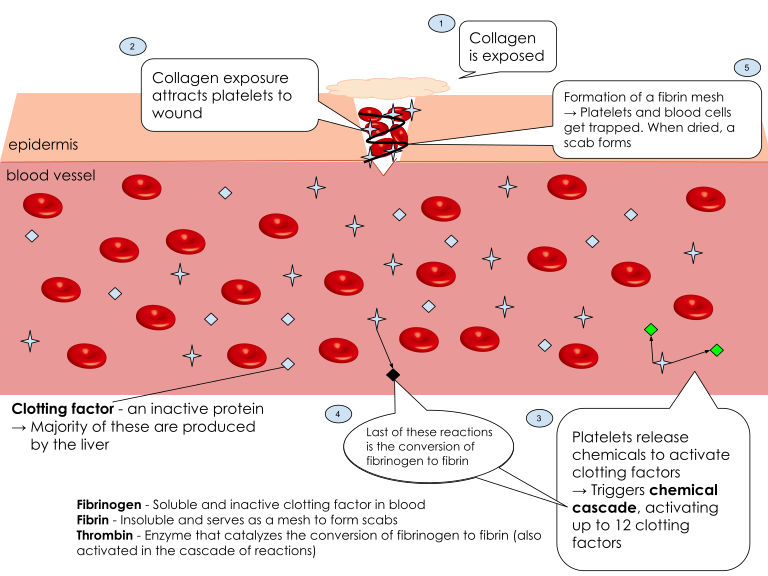
Vasokonstriksjon – kroppens første respons
Allerede før blodplater har rukket å feste seg til skaden, trekker glatte muskelceller rundt blodåren seg sammen. Dette kalles vasokonstriksjon eller vasospasme, og har som formål å redusere blodstrømmen gjennom det skadede området – slik at lekkasjen blir mindre og hemostasen får tid til å tre i kraft. Denne kontraksjonen skyldes en kombinasjon av tre mekanismer:
- Lokal utskillelse av vasokontriktorer fra skadet endotel og blodplater (som serotonin og TXA₂)
- Nevrogen refleksrespons via sensoriske nerver som reagerer på vevsskade
- Myogen kontraksjon i glatte muskelceller som strekkes
Samtidig som det skjer vasokonstriksjon i det skadede karet, kan det oppstå vasodilatasjon i nærliggende, uskadde kar, særlig ved infeksjon eller inflammasjon. Dette tillater økt tilstrømning av immunceller. Den fysiologiske differensieringen mellom innsnevring i skadet område og utvidelse i nærliggende vev viser hvor presist dette systemet er regulert.
Plateadhesjon
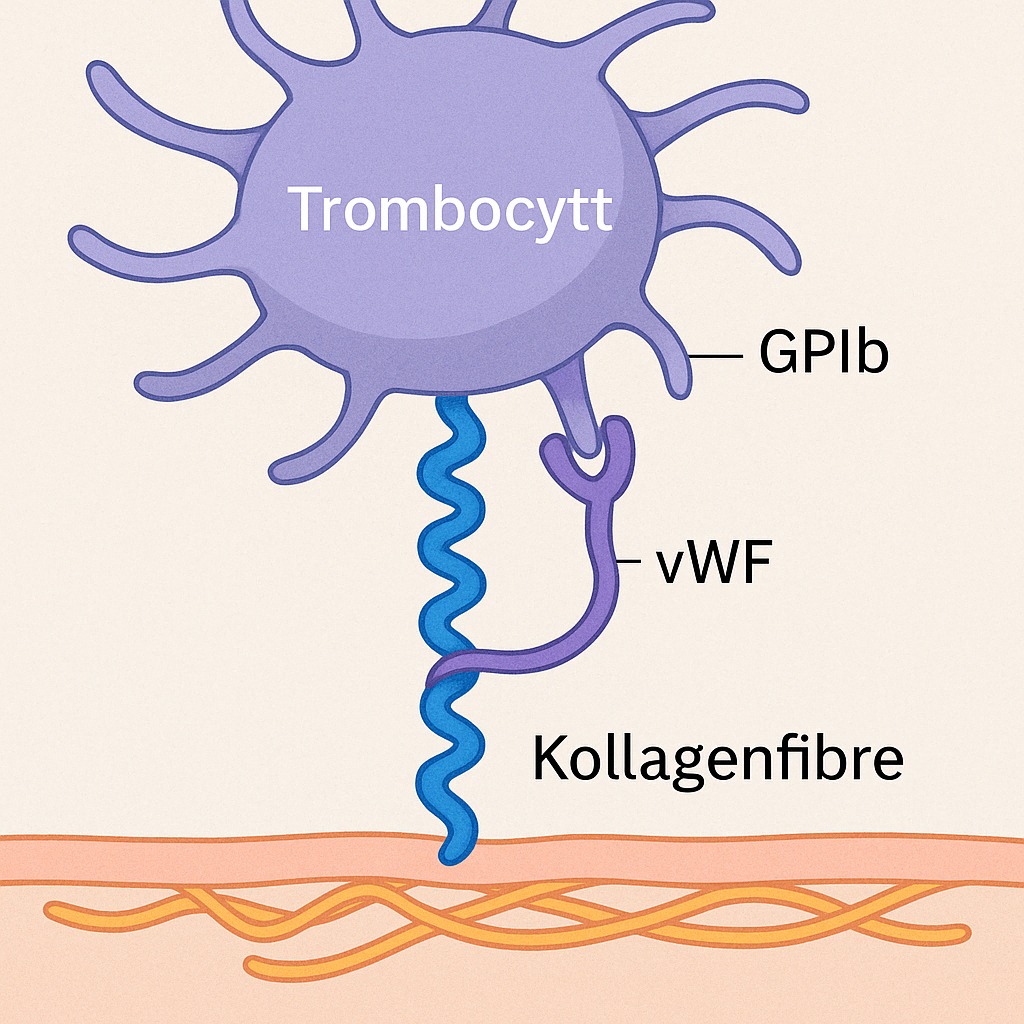
Den første forsvarslinjen i selve hemostasen er trombocyttene, også kalt blodplater. Disse små cellefragmentene sirkulerer kontinuerlig i blodet og er alltid beredt. Når de møter områder med eksponert kollagen – typisk i subendotelet – fester de seg straks. Dette kalles plateadhesjon.
Blodplatene trenger hjelp for å feste seg. Von Willebrand-faktor (vWF) fungerer som en bro mellom kollagenet og reseptoren GPIb på blodplatenes overflate (mer presist GPIb-IX-V-komplekset, da GPIb er en del av det). Dette binder seg på kollagen i den ødelagte karveggen.
Uten vWF ville trombocyttene hatt problemer med å oppdage skaden.
Dette forklarer hvorfor personer med von Willebrands sykdom har økt blødningstendens – deres “lim” mellom skaden og blodplatene fungerer dårlig eller mangler helt.
Plateaktivering og signalutskillelse
Etter at blodplatene har festet seg til kollagen via von Willebrand-faktor, aktiveres de. Aktivert trombocytt betyr at celleformen endres – den får tentakellignende utløpere og begynner en kompleks biokjemisk respons som gjør cellen klar til å forsterke hemostasen.
Biokjemisk aktivering: Fosfolipase A₂ og arakidonsyre
En del av aktiveringsprosessen innebærer at enzymene fosfolipase A₂ (PLA₂) og fosfolipase C (PLC) aktiveres. Dette fører til at fosfolipider i trombocyttens membran brytes ned og frigjør arakidonsyre, en flerumettet fettsyre som fungerer som råmateriale for flere viktige signalmolekyler.
Arakidonsyre kan deretter metaboliseres videre via to hovedveier:
- Via COX-1 (syklooksygenase-1) → danner prostaglandiner og tromboxan A₂ (TXA₂)
- Via lipoksygenase (5-LOX) → danner leukotriener, som er mer relevant for betennelsesprosesser
Blant disse stoffene er det særlig tromboxan A₂ som spiller en nøkkelrolle i hemostasen.
Degranulering og signalstoffer
Når trombocytten aktiveres, endres formen, og den starter en prosess kalt degranulering, hvor den slipper ut signalstoffer fra små lagringsblærer (granula). Disse stoffene virker både på trombocytten selv og på omkringliggende celler, og forsterker dermed plateaktiveringen.
To av de viktigste stoffene – ADP og serotonin (5-HT) – ligger ferdig lagret i granula og slippes ut umiddelbart. ADP bidrar til å rekruttere og aktivere flere trombocytter, mens serotonin fremmer lokal vasokonstriksjon.
Tromboxan A₂ (TXA₂), derimot, lagres ikke, men produseres i cytoplasmaet når trombocytten aktiveres. Fosfolipase A₂ frigjør da arakidonsyre fra cellemembranen, som videre omdannes til TXA₂ via enzymet COX-1. TXA₂ fungerer som et kraftig signalstoff som både forsterker plateaktivering og stimulerer karspasme.
Denne kombinasjonen av lagrede og nydannede signalstoffer skaper en effektiv positiv feedback-loop, som raskt bygger opp en tett og funksjonell plateplugg.
En annen viktig og ofte undervurdert endring under plateaktivering skjer i selve cellemembranen. Fosfatidylserin, et fosfolipid som normalt er lokalisert på innsiden av membranen, flyttes til utsiden i en prosess kjent som “flip-flop”.
Dette gir trombocytten en negativt ladet overflate, også kalt en trombogen membran.
Denne negativt ladede overflaten er helt avgjørende for neste steg i hemostasen – koagulasjonskaskaden. Flere viktige enzymkomplekser binder seg til fosfatidylserin via kalsiumioner, og fungerer langt mer effektivt på denne plattformen. Blant de viktigste er:
- Tenasekomplekset (faktor IXa og VIIIa)
- Protrombinasekomplekset (faktor Xa og Va)
→ Uten denne membranendringen ville ikke sekundær hemostase kunne forløpt normalt.
Plateaktivering er derfor en omfattende og irreversibel prosess, som knytter sammen den første forsvarslinjen – platepluggen – med det neste steget: fibrinforsterkningen i koagulasjonen.
Plateaggregasjon
Når nok trombocytter er aktivert, begynner de å feste seg til hverandre i en prosess som kalles plateaggregasjon.
De bruker reseptoren GPIIb/IIIa (som er et integrin), som binder til fibrinogen, og danner dermed broer mellom platene.
Resultatet er en tett, sammenklistret masse av trombocytter – en plateplugg.
Denne fungerer som et midlertidig plaster som kan stanse mindre blødninger på egen hånd.
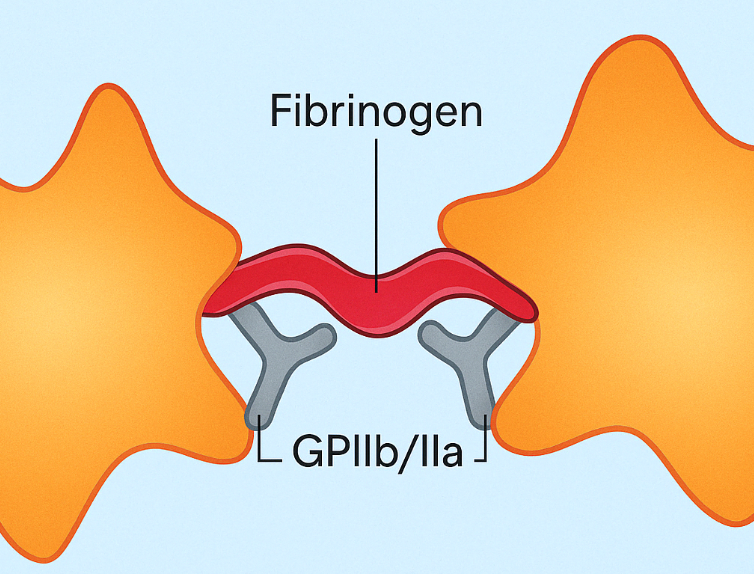
Bildet viser:
🔹 To aktiverte trombocytter (blodplater)
De har endret form – typisk for aktiverte trombocytter.
🔹 GPIIb/IIIa-reseptorer på overflaten
Disse er aktivert og tilgjengelige for binding.
🔹 Fibrinogen binder seg til GPIIb/IIIa på begge trombocyttene
→ Fungerer som en bro mellom dem
→ Dette er plateaggregasjon – altså det som danner platepluggen.
Hemming utenfor skadestedet
Systemet er imidlertid strengt regulert. Vi omgir oss med blodårer hele tiden, og trombocyttene må vite hvor de skal reagere – og hvor de ikke skal. Friske endotelceller produserer derfor prostasylkin (PGI₂) og nitrogenoksid (NO), som begge virker hemmende på trombocyttenes aktivering og aggregering. På denne måten hindres dannelsen av uønskede plugger i intakte kar.
Oversikt: Stegene i primær hemostase
- Skade på blodåre
→ Endotelet brytes, og subendotelialt kollagen eksponeres. - Plateadhesjon
→ Trombocytter fester seg til kollagen via von Willebrand-faktor (vWF) og GPIb-reseptor. - Plateaktivering
→ Trombocytter endrer form og slipper ut signalstoffer:
ADP, TXA₂, serotonin - Plateaggregasjon
→ Flere trombocytter rekrutteres og bindes sammen via
GPIIb/IIIa-reseptor og fibrinogen. - Plateplugg dannes
→ Midlertidig tetting av skadestedet. - Hemning i friskt vev
→ Uskadd endotel skiller ut NO og prostasylkin (PGI₂)
→ Hindrer plateaktivering der det ikke er skade.
Sekundær hemostase
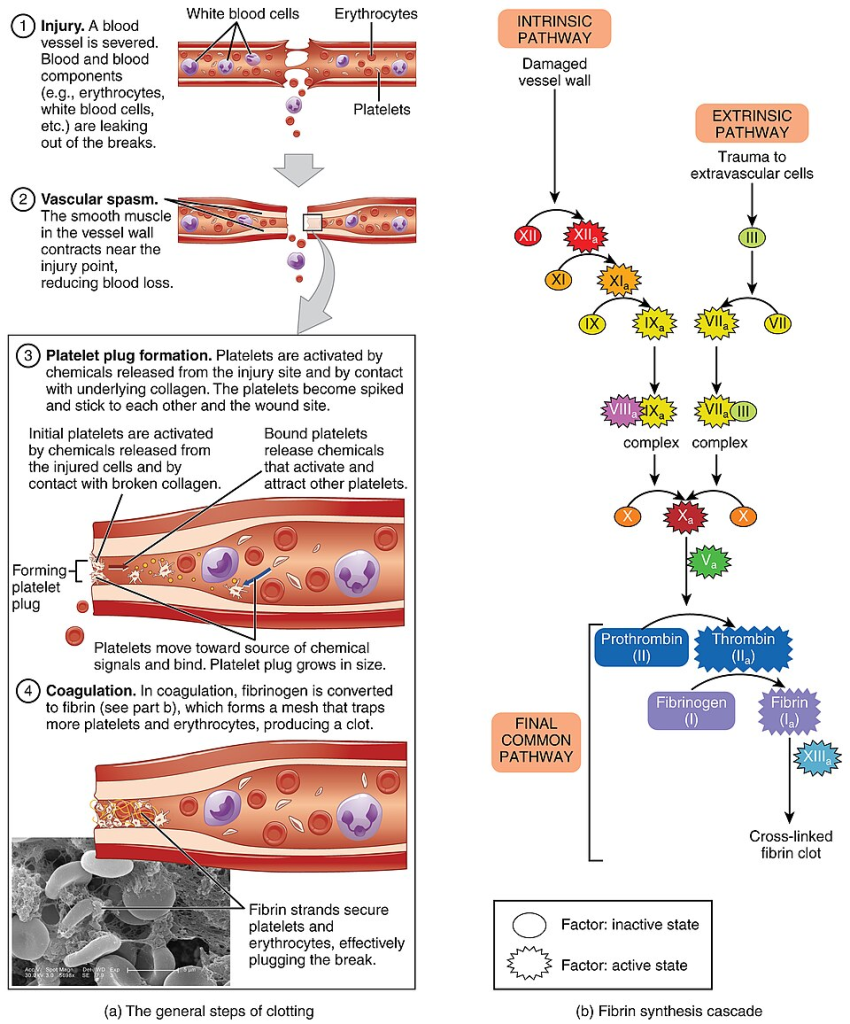
Selv om platepluggen dannet i primær hemostase raskt kan stanse en mindre blødning, er den ikke spesielt robust. Ved skade i større blodårer eller i områder med høy blodstrøm, er en midlertidig plugg av trombocytter ikke nok. Kroppen trenger en mer holdbar løsning. Det er her sekundær hemostase kommer inn: en serie enzymatiske aktiveringer som ender i dannelsen av et fibrinnettverk som forsterker og stabiliserer platepluggen.
Denne prosessen kalles koagulasjonskaskaden, og den består av en rekke koagulasjonsfaktorer – inaktive proteiner som sirkulerer i blodet og aktiveres én etter én ved at en liten del av dem klippes av. Denne formen for aktivering kalles proteolytisk aktivering, og hvert aktive enzym aktiverer det neste i rekken.
Når én faktor aktiveres, forsterkes signalet – og dette gir mulighet for rask og massiv respons med svært liten startimpuls. Det er en klassisk kaskade.
Koagulasjonskaskaden deles gjerne inn i tre deler:
- Den ekstrinsiske veien
- Den intrinsiske veien
- Den felles (common) veien
Disse tre veiene løper sammen og munner ut i dannelsen av trombin, som igjen gjør om fibrinogen til fibrin – det sentrale proteinet i koagelet.
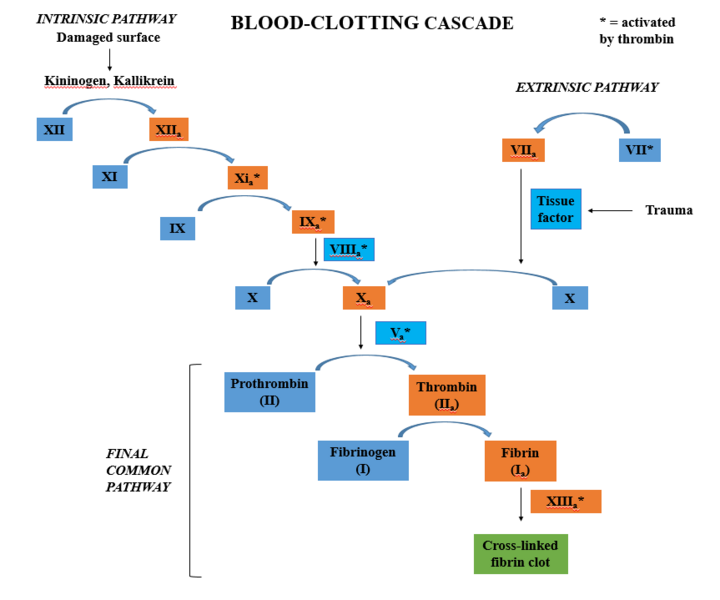
Den ekstrinsiske veien – den raskeste
Greit å vite først: I koagulasjonssystemet betyr bokstaven «a» bak faktornavnet at faktoren er aktivert – altså proteolytisk kløyvet og nå funksjonell.
Den ekstrinsiske veien starter utenfor blodbanen, altså ekstravaskulært.
Når en blodåre skades, eksponeres underliggende celler som inneholder vevsfaktor (tissue factor, TF), også kalt faktor III.
Denne finnes blant annet i membranen til glatte muskelceller.
I blodet finnes faktor VII, og en liten andel av dette er alltid i aktivert form (VIIa). Når skade skjer, binder faktor VIIa seg til vevsfaktor(faktor III) og kalsiumioner (Ca²⁺), og danner et enzymkompleks på celleoverflaten.
Dette komplekset aktiverer faktor X til faktor Xa.
Denne veien er rask og reagerer umiddelbart på skade. Den er også grunnlaget for blodprøven prothrombintid (PT) og INR.
SUPERTEIT historie for å huske det
Utenfor blodbanen lusker Agent 007 – han er selvsagt faktor VII.
Han nyser plutselig kraftig ned i tre lommetørkler – det er tissue factor (TF), altså faktor III.
Men det er ikke nok med bare spion og 3 tørkler –
han trenger et kalsiumion (Ca²⁺) og en cellemembran å jobbe på.
Disse danner et enzymkompleks sammen: TF-VIIa-komplekset, som raskt aktiverer faktor X til Xa.
Og da hopper agent 007 på det felles toget videre – til common pathway.

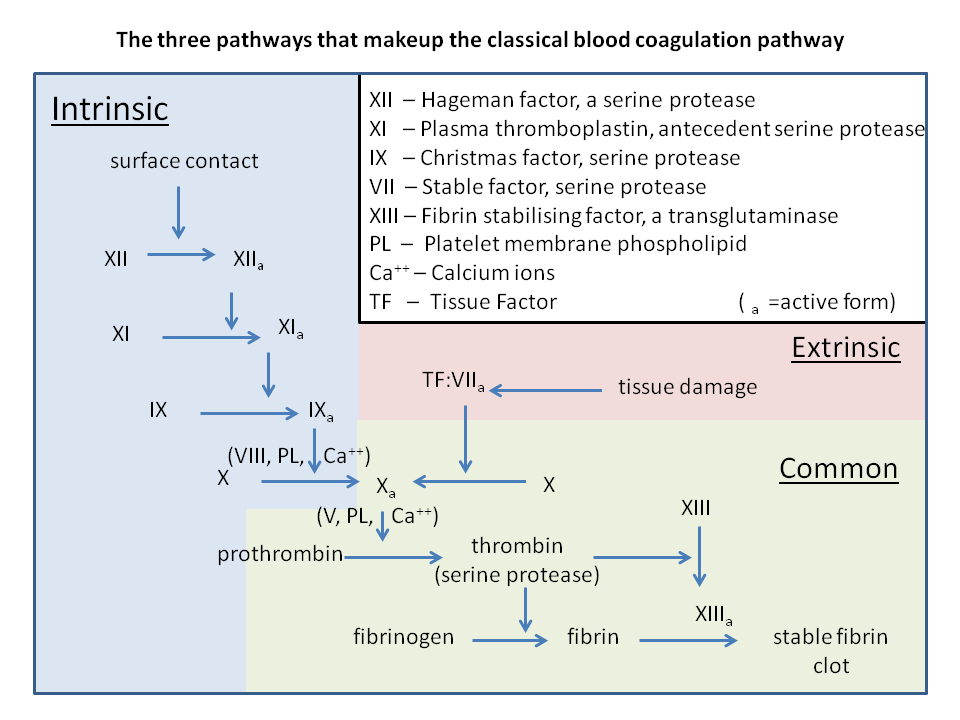
Den intrinsiske veien
Den intrinsiske veien utløses inne i blodbanen, når blod kommer i kontakt med negativt ladede overflater – som eksponert kollagen i en skadet åre eller fosfatidylserin på overflaten av aktiverte trombocytter.
Denne kontakten aktiverer faktor XII → XIIa, som deretter aktiverer faktor XI → XIa, og videre faktor IX → IXa.
Faktor IXa trenger en medhjelper – faktor VIIIa (den aktiverte formen av faktor VIII). Sammen med kalsium (Ca²⁺) og en negativt ladet fosfolipidflate (som den på aktiverte trombocytter), danner de det såkalte tenasekomplekset. (tenase betyr bokstaveligtalt: det som aktiverer faktor X, altså 10. Så lett å huske)
🔹 Tenasekomplekset = faktor IXa + faktor VIIIa + Ca²⁺ + fosfolipider
Dette komplekset aktiverer faktor X → Xa, og med det går vi over i common pathway, den felles delen av koagulasjonskaskaden for både den ekstrinsiske og den intrinsiske veien.
Faktor VIII er et ustabilt protein som raskt brytes ned i blodet, og trenger derfor beskyttelse fra von Willebrand-faktor (vWF), som frigjøres under primær hemostase. Dette er også grunnen til at pasienter med hemofili A, som mangler faktor VIII, får alvorlige blødningstendenser – særlig inn i ledd og muskler.
Den intrinsiske veien kan testes med aPTT (activated Partial Thromboplastin Time).
SUPERTEIT historie for å huske intrinsisk vei
Inne i blodbanen går en bonde rolig hjemover.
Han bærer en kurv med 12 egg – det er faktor XII.
Men så mister han ett egg! Nå har han bare 11 igjen – det er faktor XI.
Mens han bøyer seg ned for å plukke opp egget, hopper en svart katt med 9 liv ut i veien – faktor IX.
Bonden blir skremt og kaster støvelen på katten – katten overlever, men har nå bare 8 liv igjen – faktor VIII.
Men i stedet for å løpe, hopper katten opp på en plattform laget av fosfolipider og roper:
«Kom igjen, bonde! Vi bygger noe sammen!»
Bonden klatrer opp, men snubler i en blank kalsiumstein (Ca²⁺) som ligger på plattformen.
Likevel får han kastet egget videre. De bygger et kompleks av den avdøde katten med 9 liv, katten med 8 liv, litt kalsium og fosfolipider) – tenasekomplekset.
(faktor IXa + faktor VIIIa + Ca²⁺ + fosfolipider).
Dette spesielle byggverket aktiverer faktor X – og dermed er de klar for det store felles toget videre i koagulasjonskaskaden.

Felles vei
Både den ekstrinsiske og intrinsiske veien møtes i aktiveringen av faktor X til Xa. Herfra følger begge veiene den samme løypa, som leder til den faktiske dannelsen av blodproppen:
Faktor Xa aktiverer faktor V til Va. Sammen med kalsiumioner (Ca²⁺) danner disse enzymkomplekset protrombinase.
Protrombinase-komplekset har én hovedoppgave: å omdanne protrombin (faktor II) til trombin (IIa) – og dette skjer i stor skala.
Trombinproduksjonen eksploderer på dette punktet. Det er ikke snakk om ett eller to molekyler, men en trombinstorm som raskt setter i gang flere avgjørende trinn, blant annet aktivering av mer trombocytter og dannelse av fibrin! Dette er det trombin gjør:
Fibrinogen → fibrin
Trombin kløyver fibrinogen, et løselig protein som finnes i plasma, og gjør det om til fibrin, som er uløselig.
Dette danner et nettverk som holder blodplatene sammen og fanger røde blodceller, slik at proppen blir sterk og tett.
Aktivering av faktor XIII → XIIIa som kryssbinder fibrintrådene.
Dette gjør nettverket enda sterkere og mer stabilt, som lim mellom trådene.
Uten denne kryssbindingen ville fibrinet vært løst og lett oppløst.
- Aktivering av faktor V, VIII og XI (som forsterker hele prosessen, aktiverer fler faktorer fra tidligere i kaskaden)
- Aktivering av trombocytter(blodplater), som videre støtter platepluggen. Disse endrer form, eksponerer fosfatidylserin og frigjør flere signalstoffer. Det gjør at enda flere plater binder seg til proppen, og at flere koagulasjonsfaktorer aktiveres på deres overflate.
Med andre ord: dette er punktet der kroppen virkelig skrur opp volumet og forvandler en løs plateplugg til en stabil, fibrinforsterket blodpropp
SUPERTEIT historie for å huske felles vei
Den felles veien – 10-åringen, eggekremen og trombinkaka
Når både agent 007 (ekstrinsisk) og bonden med katten (intrinsisk) har gjort sin jobb, møtes de ved et kjøkken i midten av dalen. Her står en 10 år gammel baker – det er faktor X. Men han er ikke en vanlig gutt. Han er akkurat blitt aktivert, så nå kalles han faktor Xa, og han er klar til å bake koagulasjonskaka som skal tette blødningen.
Men det er ikke så enkelt å bake alene. Han roper på sin beste venn – en entusiastisk bakerhjelper som heter femmer’n (faktor V). Når femmer’n kommer, blir han straks aktivert og heter nå faktor Va.
De to begynner å sette sammen ingrediensene:
- Fem egg (symbol for faktor V)
- Litt kalsium (Ca²⁺) – det glitrer i små glass
- En stor bolle av fosfolipider – det er kjøkkenbenken deres, som egentlig er en trombocyttoverflate
Sammen visper de alt dette sammen til en mektig rørebøtte kalt protrombinasekomplekset.
Og hva skal de lage?
Jo, de heller oppi en tykk, blekgul røre kalt protrombin (faktor II). Det ser stille og rolig ut, men så fort røra kommer i kontakt med vispene – altså komplekset – skjer det noe voldsomt:
BOOM! Ut skyter trombin (IIa) – som en sprudlende bakemester med stjernestøv og energi. Han spretter ut av bollen og begynner å dekorere hele kjøkkenet i rekordfart.
Første oppdrag: Han tar tak i en stor sekk med fibrinogen (faktor I) og klipper det til fibrintråder – som sukkerpasta. Disse trådene slynges over hele kaka, limer sammen platene og bygger et sterkt nett over såret.
Andre oppdrag: Han roper på XIII – en liten limmester – og forvandler ham til XIIIa, som straks begynner å krysslime fibrintrådene, så de holder seg fast og tåler blodstrømmen.
Tredje oppdrag: Trombin smetter tilbake inn i prosessen og trykker på «forsterk»-knappen. Han gir nytt liv til faktor V, VIII og XI, så hele prosessen får et ekstra gir – alt går raskere, kraftigere og mer effektivt.
Fjerde oppdrag: Trombin trommer på bordet, og trombocyttene kommer dansende inn. De strekker ut tentakler, klemmer seg fast og forsterker platepluggen med alt de har.
Til slutt ser vi resultatet: en sterk, limt og stabil hemostatisk blodpropp – bakt og limt av en 10-åring, en femmer, noen kalsiumkuler, en trombinbaker og tonnevis av fibrintråder.

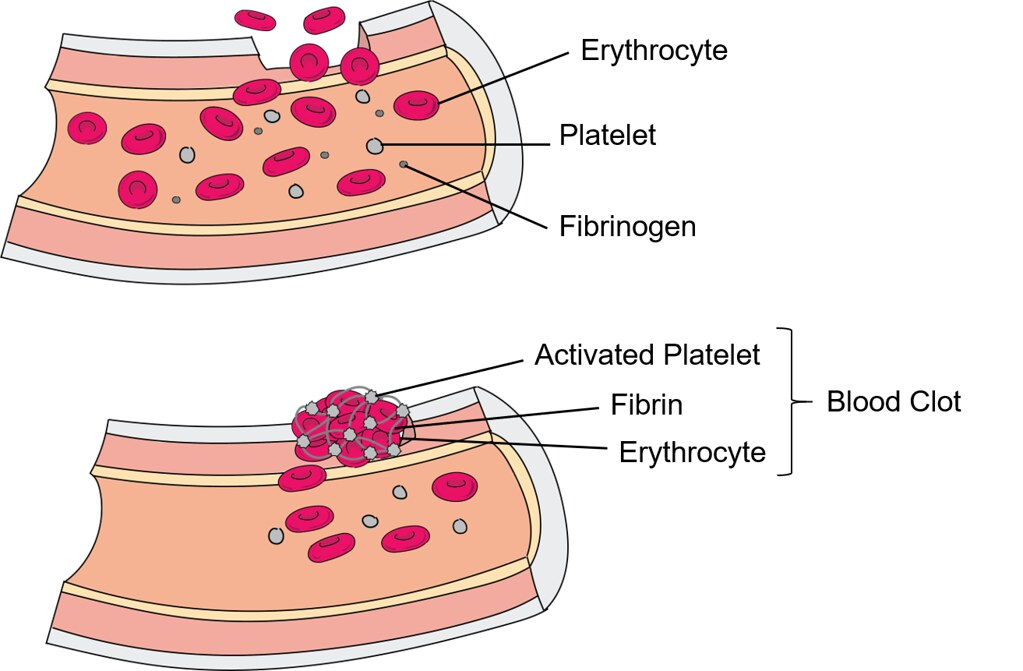
Fibrin: det som holder alt sammen
Når trombin omdanner fibrinogen til fibrin, skjer det en fysisk forandring: fibrin danner lange, uløselige tråder som fester seg til og rundt platepluggen. Sammen med røde blodceller og trombocytter danner fibrin et stabilt koagel – en blodpropp som fysisk tetter skadestedet.
Fibrinnettverket kryssbindes av faktor XIIIa og Ca²⁺, som gjør koagelet sterkere og mer motstandsdyktig mot trykket fra blodstrømmen.
Fibrinolyse
Når skaden er reparert, og blodproppen har gjort jobben sin, trenger kroppen en måte å fjerne den igjen. Det er ikke meningen at en propp skal bli værende for alltid – da risikerer vi jo å tette blodårene unødvendig. Fibrinolyse er kroppens egen, innebygde “opprydningsprosess” som sørger for å løse opp proppen når den ikke trengs mer.
I blodet finnes det et inaktivt stoff som heter plasminogen. Det ligger der som en slags soveagent, og venter på beskjed. Når endotelcellene (de som kler innsiden av blodårene) mener at “nå er det trygt, nå trenger vi ikke denne proppen mer”, skiller de ut noen hjelpere: tPA og uPA. Disse er forkortelser for “tissue plasminogen activator” og “urokinase plasminogen activator”.
Disse aktivatorene vekker plasminogen – og gjør det om til plasmin. Plasmin er et enzym som begynner å klippe opp fibrin-nettet som holder proppen sammen. Bitene som oppstår kalles fibrinnedbrytningsprodukter (forkortes FDP), og en av dem er D-dimer, som vi faktisk kan måle i blodprøver for å se om det har vært en propp i kroppen.
Fibrinolyse går ganske langsomt – og det er helt med vilje. Kroppen vil nemlig ikke løse opp proppen for tidlig. Den venter til vevet er i orden og blodsirkulasjonen er stabil igjen.
Endotelet
På innsiden av alle blodårer finner vi et tynt, men livsviktig cellelag: endotelet. Det kan ved første øyekast se passivt ut, men i virkeligheten er det en aktiv beslutningstaker i koagulasjonssystemet. Endotelet avgjør til enhver tid om det er på tide å lage en blodpropp – eller om blodet skal fortsette å flyte fritt.
Når alt er i orden – friskt endotel
I en frisk blodåre jobber endotelet aktivt for å hindre blodet i å koagulere. Det produserer og skiller ut flere hemmende signalstoffer:
- Nitrogenoksid (NO) og prostasyklin (PGI₂) virker begge karutvidende (vasodilaterende) og hemmer aktivering av trombocytter. Dette gjør at blodet flyter lettere og at blodplatene holder seg i ro.
- CD39 er et enzym på endotelets overflate som bryter ned ADP – et stoff som ellers ville stimulert trombocyttene.
- Trombomodulin binder seg til trombin og gjør trombin ufarlig – i stedet for å omdanne fibrinogen til fibrin, aktiverer det nå protein C, som hemmer koagulasjonen.
- Endotelet skiller også ut tPA (vevspasminogenaktivator), som starter fibrinolyse – altså oppløsning av blodpropp.
Med andre ord: så lenge blodkaret er intakt, sender endotelet tydelige signaler om å ikke danne blodpropp.
Når endotelet blir skadet – trombogen overflate
Så snart det skjer en skade, for eksempel ved et kutt, endrer endotelcellene karakter. De slutter å sende ut hemmende signaler – og begynner i stedet å legge til rette for koagulasjon:
- Produksjonen av NO og PGI₂ avtar → blodåren trekker seg sammen (vasokonstriksjon), og trombocyttene blir lettere aktivert.
- CD39 forsvinner, og ADP-nivåene øker → mer trombocyttaktivering.
- Produksjonen av trombomodulin reduseres → trombin virker sterkere og får danne fibrin.
- Det viktigste: endotelcellene begynner å produsere vevsfaktor (TF) som starter den ekstrinsiske veien i sekundær hemostase.
- tPA-produksjonen minker, slik at proppen ikke brytes ned for tidlig.
Resultatet er en lokalt trombogen flate – perfekt for å sette i gang både primær og sekundær hemostase akkurat der det trengs.
Trombocyttdefekter
Trombocyttene er avgjørende for den primære hemostasen – altså for dannelsen av den første platepluggen. Hvis blodplatene ikke virker normalt, kan man få blødningstendenser, særlig i slimhinner, hud og ved menstruasjon. Dette kalles umiddelbar hemostasesvikt, og skyldes ofte defekter i blodplatenes evne til å feste seg (adhesjon) eller binde seg sammen (aggregasjon).
Vanlige og sjeldne årsaker:
- Von Willebrands sykdom (vanlig): Skyldes mangel på eller dysfunksjon av von Willebrand-faktor. Dette gir problemer med plateadhesjon og stabilisering av faktor VIII.
- Glanzmanns trombasteni (sjeldnere): Skyldes en defekt i GPIIb/IIIa-reseptoren, som er nødvendig for at blodplatene skal kunne binde seg sammen via fibrinogen.
- Bernard-Souillers syndrom (meget sjelden): Skyldes defekt i GPIb/IX-komplekset, som trengs for at blodplatene skal feste seg til kollagen via vWF.
Medikamentell behandling/farmakologi
Protein C og protein S – kroppens egne koagulasjonsbremser
Mens flere legemidler kan hemme koagulasjonskaskaden medikamentelt, finnes det også naturlige bremsemekanismer i kroppen. De viktigste av disse er protein C og protein S. Disse er ikke koagulasjonsfaktorer i klassisk forstand, men naturlige antikoagulantia som spiller en avgjørende rolle i å holde koagulasjonen i balanse.
Protein C aktiveres til aktivert protein C (APC) ved hjelp av trombin – men bare når trombin er bundet til et spesialisert overflateprotein kalt trombomodulin, som finnes på endotelceller. Når APC er aktivert, fungerer det som en «saks» og klipper bort to viktige koagulasjonsforsterkere: faktor Va og faktor VIIIa.
På den måten reduserer APC aktiviteten i både den indre og den felles veien av koagulasjonskaskaden.
Protein S fungerer som en kofaktor for aktivert protein C, og hjelper til med å stabilisere og forsterke virkningen av APC. Sammen danner de et viktig forsvar mot ukontrollert koagulasjon og trombose.
Dersom protein C eller S mangler, enten genetisk eller som følge av sykdom (for eksempel ved alvorlig infeksjon, DIC eller leversvikt), øker risikoen for blodpropp betydelig. Disse manglene er derfor viktige å vurdere ved uforklarlig venøs tromboembolisme hos yngre pasienter.
Trombocytthemmere
For å redusere risikoen for blodpropp – særlig i arterier, hvor trykket og blodstrømmen er høy – brukes medikamenter som hemmer trombocyttenes funksjon. Disse kalles trombocytthemmere, og virker ved å hindre enten adhesjon (at platene fester seg) eller aggregasjon (at de klumper seg sammen).
Hemming av adhesjon
Først må trombocyttene feste seg til skadestedet. Dette skjer blant annet via GPIIb/IIIa-reseptoren, som binder fibrinogen og danner broer mellom blodplatene. Ved å blokkere denne reseptoren, kan man effektivt hemme trombocyttadhesjon. Et eksempel på et slikt legemiddel er eptifibatid, som er et antistoff som binder reseptoren og hindrer at platene kobler seg sammen.
Hemming av aggregasjon
Etter at blodplatene har festet seg, starter en kraftig aktiveringsprosess hvor de slipper ut signalstoffer. Her kan vi gripe inn på flere måter:
- COX-hemmere, som acetylsalisylsyre (ASA) og NSAIDs, hemmer enzymet COX-1, som er nødvendig for produksjon av tromboxan A₂ (TXA₂). Siden TXA₂ er et viktig aktiveringssignal, vil dette dempe plateaktiveringen og rekrutteringen av flere trombocytter.
- ADP-reseptorhemmere, som klopidogrel (Plavix) og ticagrelor (Brilique), hindrer ADP i å binde seg til trombocyttenes overflate. ADP er et signalstoff som forsterker aktiveringen og rekrutterer flere plater. Ved å blokkere reseptoren, avbrytes denne forsterkende sløyfen.
Hovedbruksområde: Arterielle tromboser – f.eks. hjerteinfarkt og hjerneslag.
Koagulasjonsfaktorhemmere
Mens trombocytthemmere forhindrer dannelsen av den første platepluggen, virker koagulasjonsfaktorhemmere på neste trinn: selve koagulasjonskaskaden – altså prosessen som fører til at fibrin dannes og danner et stabilt nettverk som fanger celler og holder pluggen sammen.
Ved å hemme denne kaskaden, forhindrer man dannelsen av fibrinproppen, og dermed reduserer man risikoen for uønskede blodpropper, særlig i venesystemet, hvor koagulasjonen spiller hovedrollen.
Vitamin K-antagonister – klassikeren (f.eks. warfarin)
Disse legemidlene hemmer enzymet som er nødvendig for at flere viktige koagulasjonsfaktorer skal modnes. Uten vitamin K får man ikke karboksylering av faktor II, VII, IX og X – og uten denne prosessen fungerer ikke faktorene.
Resultatet er en bred hemming av både:
- Den ekstrinsiske veien (faktor VII)
- Den felles veien (faktor X og II)
Warfarin krever jevnlig blodprøvekontroll (INR – internasjonal normalisert ratio) for å sikre riktig dosering og effekt, da små doseendringer kan gi stor blødnings- eller proppfare.
Direkte orale antikoagulantia (DOAKs)
Disse medikamentene virker direkte på enkeltfaktorer og trenger ikke vitamin K for å fungere. De er enklere å bruke enn warfarin og krever vanligvis ikke rutinemessig blodprøvekontroll.
De deles inn etter målpunktet i kaskaden:
- Faktor Xa-hemmere:
- Rivaroksaban, apiksaban, edoksaban
- Disse hemmer faktor Xa direkte og hindrer dermed at trombin dannes.
- Trombinhemmere:
- Dabigatran
- Hemmer trombin (faktor IIa) direkte og forhindrer at fibrinogen → fibrin.
Heparin og lavmolekylært heparin
Heparin og LMWH (f.eks. enoksaparin) virker ved å forsterke effekten av kroppens eget antikoagulerende system, nemlig antitrombin III. Dette proteinet inaktiverer både:
- Faktor Xa
- Trombin (IIa)
Ved å gjøre antitrombin III mer effektiv, bremser heparin koagulasjonen på to viktige steder.
Heparin gis alltid som injeksjon, og brukes ofte i sykehus ved:
- Akutt dyp venetrombose
- Lungeemboli
- Postoperativ profylakse
Hovedbruksområde: Venøse tromboser – f.eks. dyp venetrombose og lungeemboli.
📚 Anki-kort
Obs, tomt! Kommer etterhvert <3
📝 Eksamensoppgaver
Obs, tomt! Kommer etterhvert <3
👨⚕️ Klinisk case
Obs, tomt! Kommer etterhvert <3
❓ Test deg selv
Obs, tomt! Kommer etterhvert <3