
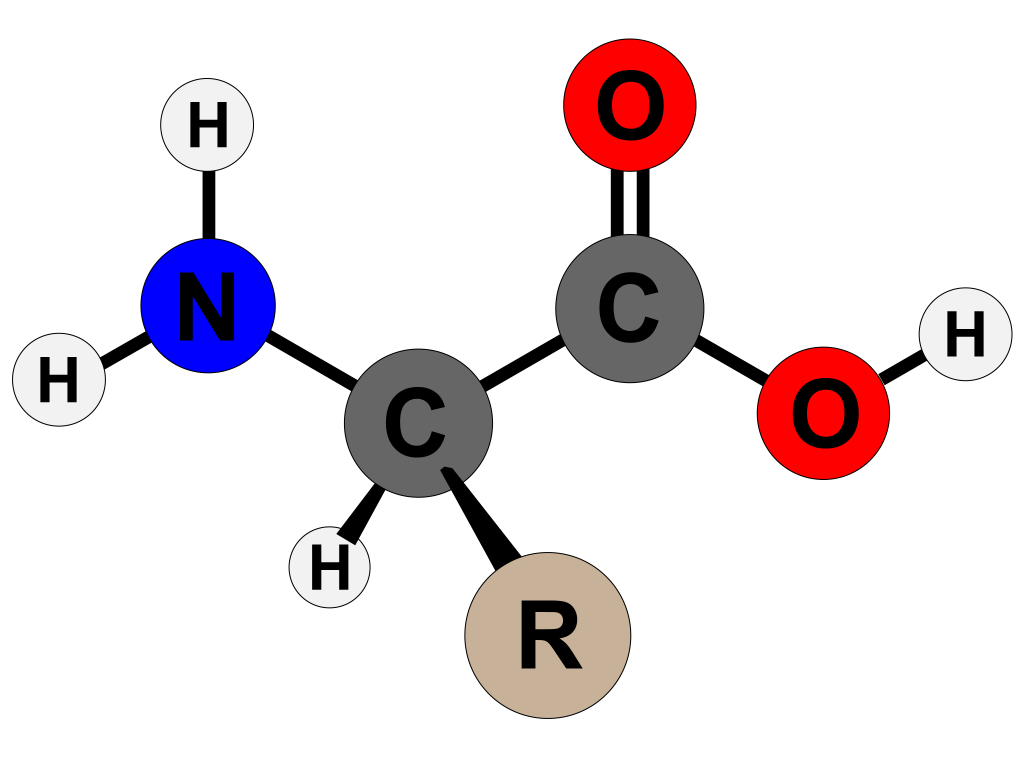
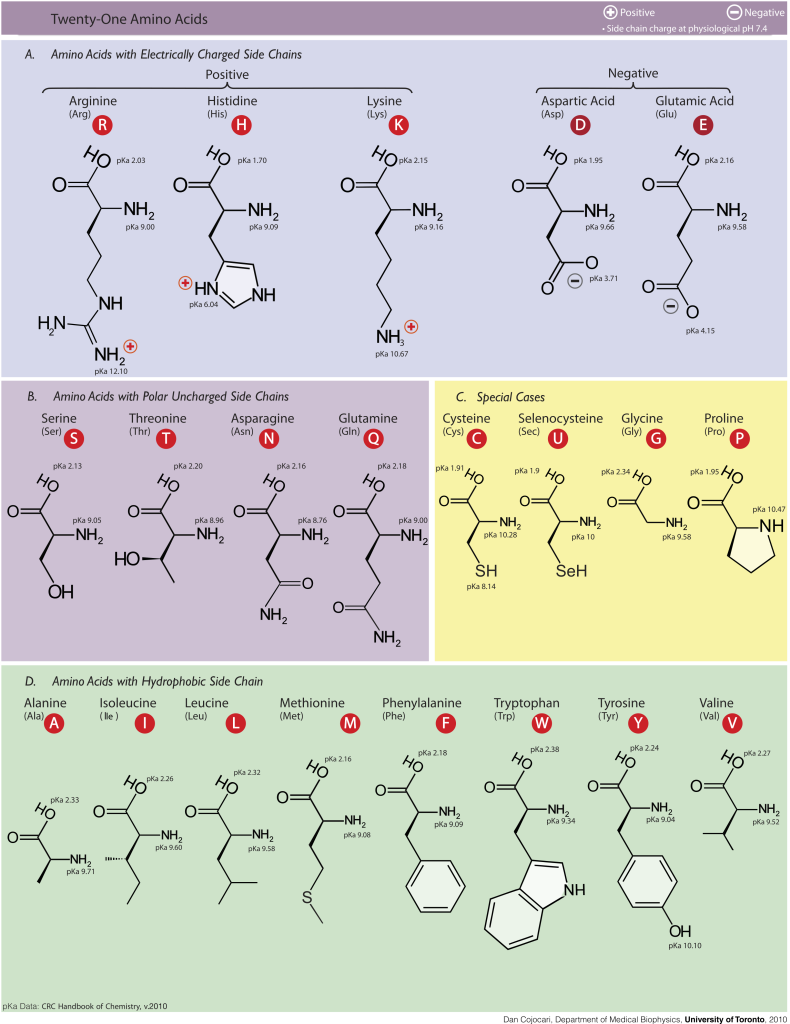
Aminosyrer, byggesteinene i proteiner, har en kompleks struktur bestående av en nitrogenholdig gruppe og et karbonskjelett. Nitrogengruppen er essensiell i metabolismen, og når aminosyrene brytes ned, blir nitrogenet i aminosyrene frigjort i form av ammoniakk. Denne prosessen er en viktig del av hvordan kroppen håndterer proteiner og andre nitrogenholdige forbindelser.
Hvordan Ammoniakk Dannes
Under nedbrytningen av aminosyrer, spesielt under transaminering, fjernes aminosyrenes nitrogenholdige gruppe. Dette skjer ved at aminogrupper (NH2) overføres fra en aminosyre til et annet molekyl. Resultatet av denne reaksjonen er dannelsen av ammoniakk (NH3), et giftig biprodukt. Ammoniakk er svært reaktivt og kan være skadelig for kroppen hvis det ikke fjernes raskt.
Toksisitet av Ammoniakk
Ammoniakk i høy konsentrasjon er ekstremt giftig, spesielt for hjernen. Det kan forstyrre normal nevrotransmisjon, noe som kan føre til alvorlige nevrologiske symptomer som forvirring, koma og til og med død dersom det ikke omdannes til et mindre giftig molekyl. Derfor er det viktig at kroppen har effektive mekanismer for å håndtere og eliminere ammoniakk.
Transport av Ammoniakk til Leveren
Ammoniakk, som dannes i forskjellige vev, fraktes til leveren for å bli omdannet til urea i ureasyklusen. Denne prosessen er avgjørende for å fjerne ammoniakk fra kroppen, da det ellers kan akkumuleres og forårsake toksisitet.
Det finnes flere mekanismer som bidrar til å transportere ammoniakk til leveren, og de involverer både glutamin og alanin, som fungerer som transportører.
Glutamin Syntetase og Glutamin som Transportør
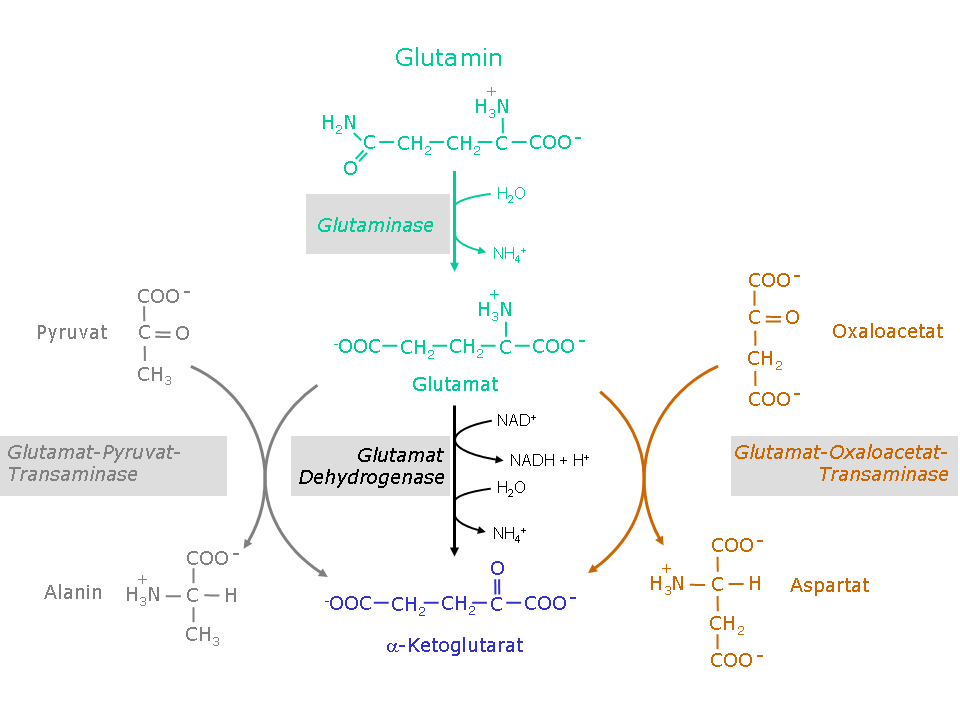
Glutamin syntetiseres i perifere vev, som muskler og hjernen, og fungerer som en viktig transportør av ammoniakk til leveren.
Glutamin dannes når glutamat reagerer med ammoniakk, katalysert av enzymet glutaminsyntetase.
Glutamin er en stabil form for ammoniakk og kan fraktes trygt gjennom blodet uten at det forårsaker skade på andre vev. Når glutamin når leveren, omdannes det tilbake til glutamat, og ammoniakk frigjøres for videre omdannelse i ureasyklusen.
Muskelens Rolle i Ammoniakktransport
Musklene spiller en nøkkelrolle i transporten av ammoniakk gjennom dannelsen av alanin. I muskelfibrene kan glutamat, som har fått overført en aminogruppe, omdannes til alanin gjennom transaminering. Denne prosessen skjer i muskelcellene, og alanin fungerer som en transportør av ammoniakk fra muskelvevet til leveren. Når alanin når leveren, kan det deamineres tilbake til glutamat, og ammoniakken som ble transportert med alanin, kan deretter bearbeides i ureasyklusen.
Enzymet som er viktig for denne prosessen er glutamat dehydrogenase, som katalyserer omdannelsen av glutamat til alfa-ketoglutarat samtidig som ammoniakk frigjøres. Denne prosessen skjer i muskelcellene og er avgjørende for å opprettholde balansen i nitrogenmetabolismen.
Alanin Transaminase (ALT)
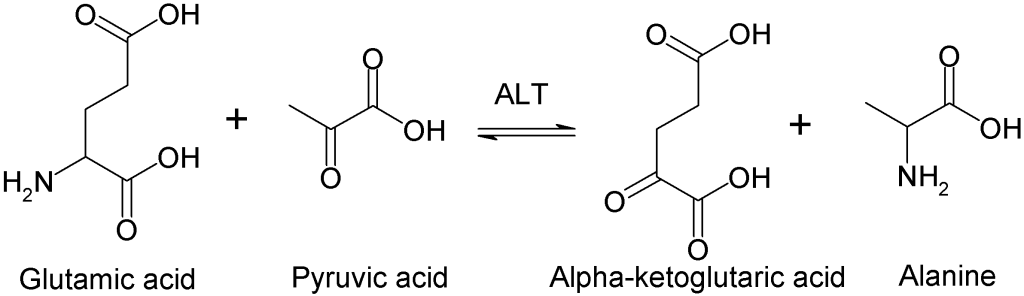
Alanin transaminase (ALT) er et viktig enzym som muliggjør transporten av ammoniakk fra muskelcellene til leveren via alanin. Enzymet katalyserer en transaminering, der aminogruppen fra glutamat overføres til pyruvat, og danner alanin. Alanin kan deretter transporteres til leveren, hvor det brukes til glukoneogenese eller blir omdannet til urea gjennom ureasyklusen.
ALT spiller derfor en kritisk rolle i den transamineringen som muliggjør at ammoniakk, som kan være toksisk, trygt transporteres til leveren, hvor det kan omdannes til urea og elimineres fra kroppen.
Nedbrytning i Levercellene
Leveren spiller en sentral rolle i nedbrytning av aminosyrer og håndtering av ammonium (NH₄⁺). Flere enzymer i leveren er involvert i prosessene som frigjør ammonium, og det omdannes til urea gjennom ureasyklusen, som skjer i både mitokondriene og cytosol.
Prosesser som Frigjør NH₄⁺:
- Glutaminase: Glutaminase omdanner glutamin til glutamat og frigjør ammonium i prosessen. Dette er en viktig mekanisme for å regulere glutaminnivåene og produserer ammonium som kan gå videre til ureasyklusen.
- Glutamat dehydrogenase: Dette enzymet katalyserer den oksidative deamineringen av glutamat, og frigjør en annen ammoniumion (NH₄⁺). Denne reaksjonen er reversibel og spiller en nøkkelrolle i nedbrytningen av aminosyrer og dannelsen av ammonium.
- Andre deaminaser: Flere enzymer, som serin dehydratase og histidase, kan også frigjøre ammonium fra andre aminosyrer, og bidrar til å regulere nitrogenbalansen i kroppen.
Bruk av NH₄⁺ i Ureasyklusen
De ammoniumionene (NH₄⁺) som frigjøres i levercellene blir transportert til ureasyklusen. Denne syklusen finner sted hovedsakelig i levers celler og omdanner giftig ammoniakk til urea, som er mye mindre toksisk. Urea kan deretter skilles ut via nyrene.
Ureasyklusen består av en serie enzymatiske reaksjoner som skjer i både mitokondriene og cytosol i leveren. Hensikten med denne syklusen er å effektivt fjerne overskudd av nitrogen fra kroppen, og gjøre det mulig å eliminere det via urin.
Urea-Syklusen
Når ammonia (NH₃) har nådd leveren, starter prosessen med å omdanne dette giftige stoffet til urea, et stoff som kroppen kan skille ut trygt via urinen.
Dette skjer gjennom en serie reaksjoner kjent som urea-syklusen. Syklusen finner sted i to deler av levercellene – i mitokondriene og i cytoplasmaet.
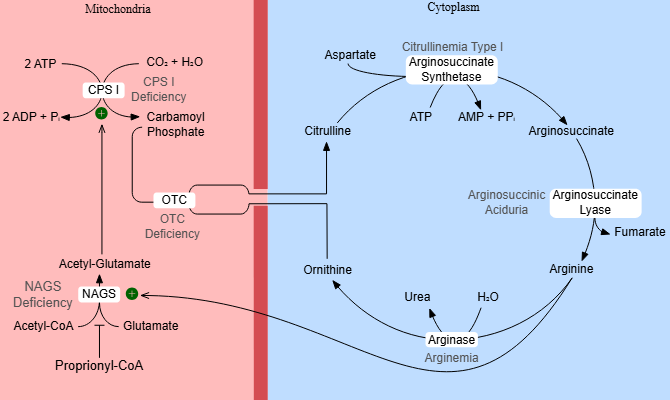
Starten på Urea-Syklusen – Dannelse av Karbamoylfosfat
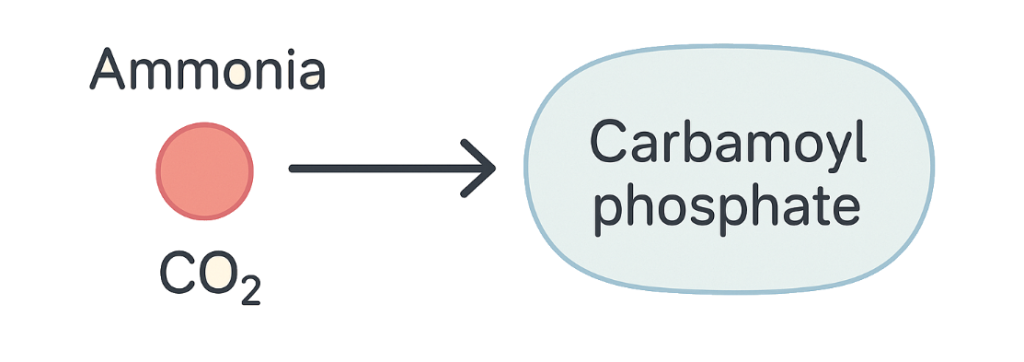
Den første reaksjonen i urea-syklusen skjer i mitokondriene.
Ammoniaen som har blitt transportert til leveren gjennom blodet, reagerer med karbondioksid (CO₂) for å danne karbamoylfosfat, et viktig mellomprodukt.
Denne reaksjonen krever energi i form av ATP, og enzymet som muliggjør denne prosessen, heter karbamoylfosfat syntetase 1 (CPS1).
CPS1 er en nøkkelkomponent i denne prosessen, og uten det ville ikke syklusen kunne starte.
En viktig aktivator for CPS1 er N-acetylglutamat. Dette stoffet binder seg til en annen del av CPS1, og gjør at enzymet kan behandle ammonia mer effektivt. Uten tilstrekkelig N-acetylglutamat i leveren kan ammonia bygge seg opp og bli giftig, noe som kan føre til alvorlige helseproblemer.
Steg 2: Fra Karbamoylfosfat til Citrullin
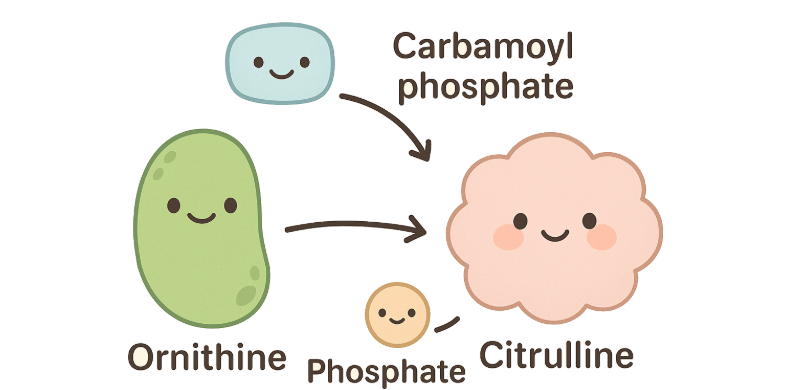
Når karbamoylfosfat er dannet, skjer neste reaksjon også i mitokondriene. Her reagerer karbamoylfosfat med ornithin, en annen aminosyre, og danner et nytt molekyl kalt citrullin.
Denne reaksjonen skjer ved hjelp av enzymet ornithin transkarbamoylase, som frigjør et fosfatmolekyl i prosessen.
Citrullin er et viktig mellomprodukt som nå kan forlate mitokondriene og bevege seg inn i cytoplasmaet, hvor den vil reagere med et annet stoff for videre bearbeiding.
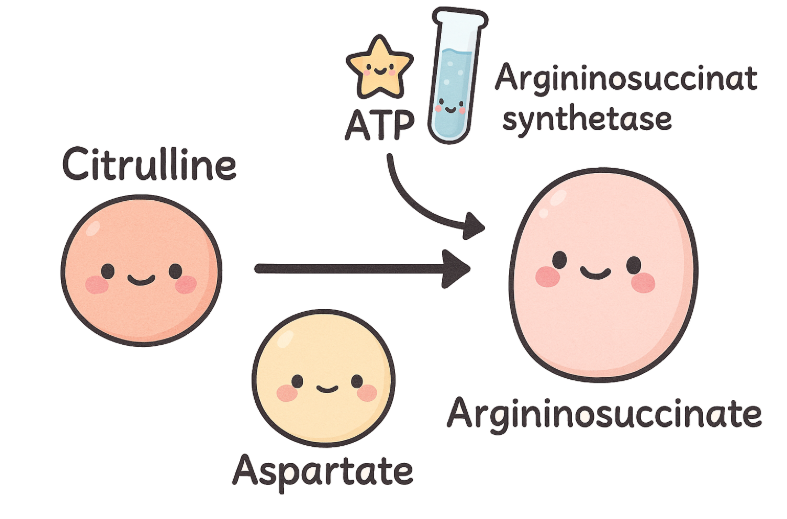
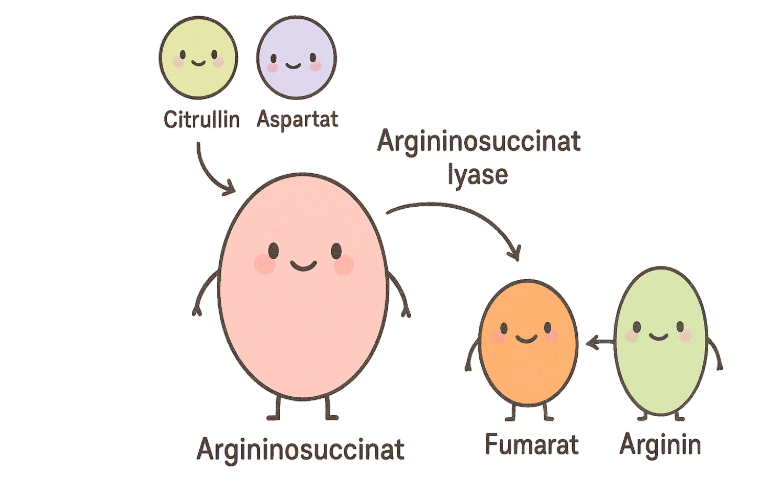
Steg 3: Fra Citrullin til Argininosuccinat – Kombinasjonen med Aspartat
I cytoplasmaet møtes citrullin med aspartat, en annen aminosyre som bidrar med et ekstra nitrogenatom. Dette nitrogenet er viktig for å bygge videre på strukturen som til slutt skal danne urea.
Reaksjonen mellom citrullin og aspartat skjer ved hjelp av enzymet argininosuccinat synthetase, og denne reaksjonen krever energi i form av ATP. Det er nå at aspartat gir sitt andre nitrogenatom til urea-syklusen.
Nå har vi dannet argininosuccinat, som inneholder begge nitrogenatomene som trengs for å lage urea.
Steg 4: Nedbrytning av Argininosuccinat – Dannelse av Fumarat og Arginin
Neste trinn i syklusen innebærer at argininosuccinat brytes ned til to produkter: fumarat og arginin. Dette skjer ved hjelp av enzymet argininosuccinat lyase.
- Fumarat, som dannes i denne prosessen, går videre til et annet stoffskifteprodukt i cellen og kan brukes videre i syklusen for å regenerere et viktig mellomprodukt.
- Arginin er det andre produktet, og det spiller en kritisk rolle i neste trinn for å fullføre urea-syklusen.
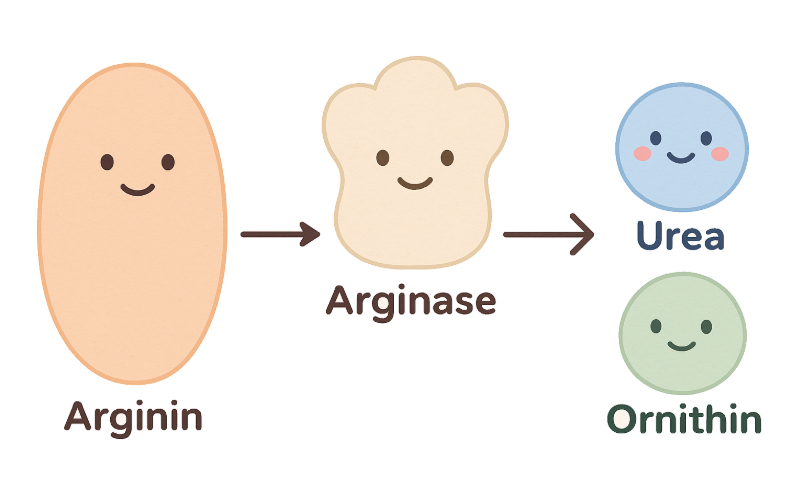
Steg 5: Fra Arginin til Urea – Den Endelige Prosessen
Argenin blir deretter videre bearbeidet av enzymet arginase, som bryter det ned til urea og ornithin.
- Urea er det endelige produktet, og det kan nå transporteres ut av leveren og inn i blodbanen, der det sendes til nyrene for utskillelse.
- Ornithin går tilbake til mitokondriene for å delta i den neste runden av syklusen.
Nå har vi effektivt omdannet den giftige ammoniaen til trygt urea, som kan skilles ut av kroppen.
Utskillelse av Urea
Når urea er dannet, blir det transportert fra leveren til blodet og derfra til nyrene. Nyrene filtrerer urea fra blodet, og det blir til slutt utskilt i urinen, som en trygg måte å fjerne giftstoffer fra kroppen på.
Regulering av ureasyklusen
For at ureasyklusen skal fungere optimalt og tilpasses kroppens stadig skiftende behov, må aktiviteten til enzymene som inngår i syklusen reguleres nøye. Reguleringen av ureasyklusen er avgjørende for at kroppen skal kunne håndtere ulike mengder ammoniakk, som varierer med proteininntak og graden av proteinnedbrytning.
Den viktigste reguleringen av ureasyklusen foregår ved det første enzymet i syklusen: karbamoyl-fosfat-syntetase 1 (CPS1). CPS1 katalyserer det aller første steget i ureasyklusen og er derfor et naturlig kontrollpunkt. CPS1 befinner seg i mitokondriene i levercellene, der det kombinerer ammoniakk med karbondioksid og ATP til å danne karbamoyl-fosfat.
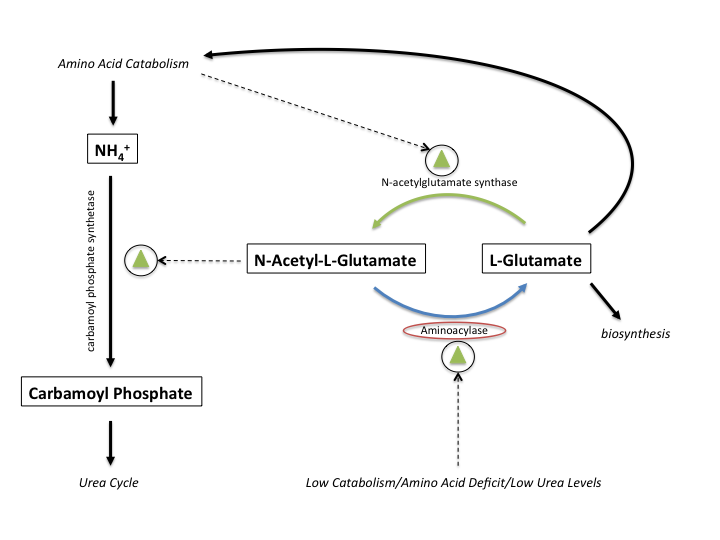
Regulering av CPS1 – rollen til N-acetylglutamat
Det sentrale reguleringsmolekylet for CPS1 er N-acetylglutamat (NAG). CPS1 er sterkt avhengig av NAG som en positiv allosterisk aktivator – det betyr at CPS1 kun fungerer effektivt dersom NAG er til stede.
N-acetylglutamat produseres av enzymet N-acetylglutamat-syntase, som igjen aktiveres av høye konsentrasjoner av aminosyren arginin. Når kroppen bryter ned mye protein, øker nivået av arginin. Økt arginin signaliserer dermed til leveren at det er behov for å fjerne mer ammoniakk. Dette gjøres gjennom at arginin stimulerer produksjonen av NAG, som igjen aktiverer CPS1 og øker hastigheten på ureasyklusen.
Forenklet forklart:
Når vi spiser mye protein eller bryter ned mye aminosyrer, øker mengden ammoniakk i kroppen. Ammoniakk må fjernes raskt, og derfor øker argininmengden. Arginin gir beskjed til enzymet N-acetylglutamat-syntase om å produsere mer N-acetylglutamat. Dermed blir CPS1 mer aktiv, og ureasyklusen jobber raskere med å fjerne ammoniakk.
Regulering på lengre sikt – endring i enzymmengder
Ved lengre tids endringer i proteininntaket kan også mengden enzymer i ureasyklusen justeres. Ved et kosthold rikt på proteiner, vil genuttrykket for ureasyklusens enzymer øke, slik at leverens kapasitet for ammoniakkdetoksifisering økes. Motsatt vil lavt proteininntak eller proteinmangel redusere mengden av disse enzymene. Denne reguleringen skjer via hormonelle og metabolske signaler, som påvirker transkripsjonen av genene som koder for enzymene i ureasyklusen.
Klinisk relevans:
Ved genetiske defekter eller svikt i reguleringen av ureasyklusen kan ammoniakknivåene stige kraftig i blodet, noe som fører til hyperammonemi – en potensielt livstruende tilstand som krever umiddelbar behandling.
Oppsummert er reguleringen av ureasyklusen en nøye balansert prosess hvor kortsiktig regulering skjer ved hjelp av arginin og N-acetylglutamat, og langsiktig regulering involverer endringer i enzymproduksjon. Denne kombinasjonen av reguleringsmekanismer sikrer effektiv avgiftning av ammoniakk og optimal tilpasning til kroppens metabolske behov.
Energiproduksjon fra Aminosyrer
Aminosyrer spiller en viktig rolle ikke bare som byggesteiner for proteiner, men også som energikilder. Når kroppen trenger energi, kan aminosyrer brytes ned til mellomprodukter som kan benyttes i forskjellige metabolske prosesser som glukoneogenese (produksjon av glukose) eller ketonlegemedannelse. Disse prosessene fører til dannelse av ATP, som er den primære energikilden i cellene.
Glukogene og Ketogene Aminosyrer
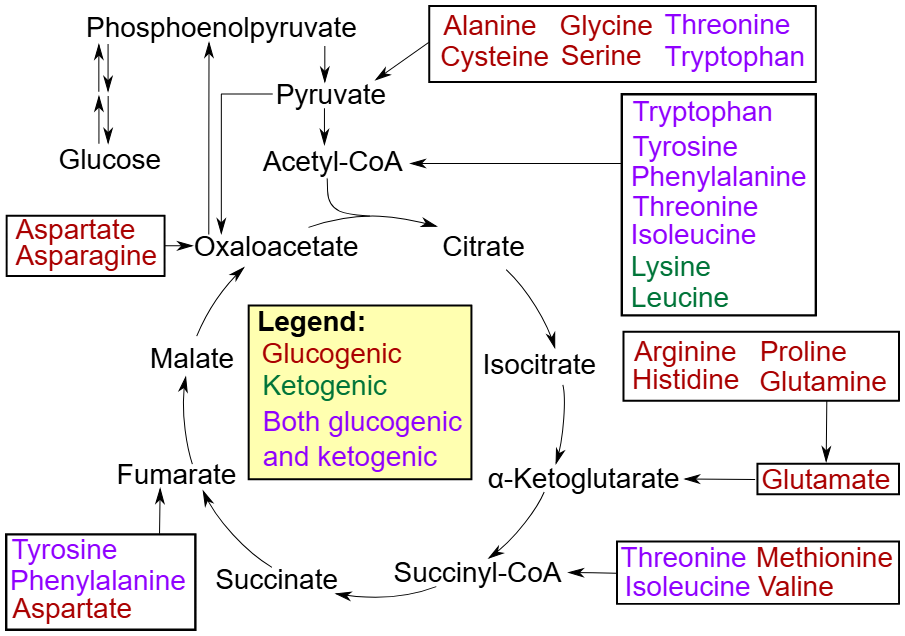
Aminosyrer kan deles inn i to kategorier: glukogene og ketogene aminosyrer, avhengig av hva de kan omdannes til i kroppen.
Glukogene aminosyrer omdannes til glukose gjennom en prosess som kalles glukoneogenese. Denne prosessen er essensiell under faste eller katabolske tilstander, som for eksempel ved sykdom eller intens fysisk aktivitet. Glukosen som produseres kan deretter brukes som energi for cellene. Eksempler på glukogene aminosyrer inkluderer serin, asparagin og glutamin.
Ketogene aminosyrer, derimot, omdannes til ketonlegemer som acetoacetat og β-hydroksybutyrat. Ketonlegemer kan fungere som alternative energikilder, spesielt når glukose er utilgjengelig, som for eksempel under en ketogen diett eller ved langvarig faste. De viktigste ketogene aminosyrene er leucin og lysin.
Spesifikke Vev og Energiomsetning
Noen aminosyrer spiller en mer spesifikk rolle i forskjellige vev og organer. For eksempel er glutamin en viktig energikilde for cellene i tarmen og immuncellene (som lymfocytter). Glutamin kan omdannes til glutamat og videre til α-ketoglutarat, et viktig mellomprodukt i sitronsyresyklusen, som er en sentral energiproduserende prosess i cellene.
Essensielle og Ikke-Essensielle Aminosyrer
Aminosyrer kan også klassifiseres som essensielle eller ikke-essensielle.
Essensielle aminosyrer er de som kroppen ikke kan produsere selv, og derfor må de tilføres gjennom kosten. Eksempler på essensielle aminosyrer er leucin, isoleucin, valin, metionin, fenylalanin, treonin, tryptofan og valin. Uten disse aminosyrene fra kosten kan kroppen ikke syntetisere de proteinene den trenger.
Ikke-essensielle aminosyrer, derimot, kan kroppen lage selv fra andre forbindelser, for eksempel gjennom transaminering av karbonskjelett fra glykolysen eller sitronsyresyklusen. Eksempler på ikke-essensielle aminosyrer er glutamat, alanyn og asparagin.
Energiproduksjon fra Aminosyrer
Aminosyrer spiller en viktig rolle ikke bare som byggesteiner for proteiner, men også som energikilder. Når kroppen trenger energi, kan aminosyrer brytes ned til mellomprodukter som kan benyttes i forskjellige metabolske prosesser som glukoneogenese (produksjon av glukose) eller ketonlegemedannelse. Disse prosessene fører til dannelse av ATP, som er den primære energikilden i cellene.
Glukogene og Ketogene Aminosyrer
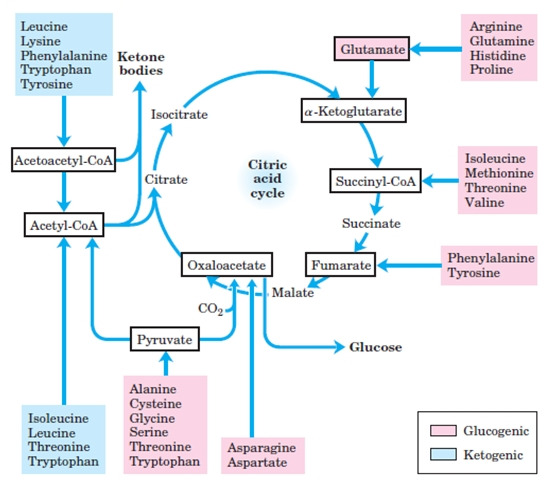
Aminosyrer kan deles inn i to kategorier: glukogene og ketogene aminosyrer, avhengig av hva de kan omdannes til i kroppen.
Glukogene aminosyrer omdannes til glukose gjennom en prosess som kalles glukoneogenese. Denne prosessen er essensiell under faste eller katabolske tilstander, som for eksempel ved sykdom eller intens fysisk aktivitet. Glukosen som produseres kan deretter brukes som energi for cellene. Eksempler på glukogene aminosyrer inkluderer serin, asparagin og glutamin.
Ketogene aminosyrer, derimot, omdannes til ketonlegemer som acetoacetat og β-hydroksybutyrat. Ketonlegemer kan fungere som alternative energikilder, spesielt når glukose er utilgjengelig, som for eksempel under en ketogen diett eller ved langvarig faste. De viktigste ketogene aminosyrene er leucin og lysin.
Spesifikke Vev og Energiomsetning
Noen aminosyrer spiller en mer spesifikk rolle i forskjellige vev og organer. For eksempel er glutamin en viktig energikilde for cellene i tarmen og immuncellene (som lymfocytter). Glutamin kan omdannes til glutamat og videre til α-ketoglutarat, et viktig mellomprodukt i sitronsyresyklusen, som er en sentral energiproduserende prosess i cellene.
Dannelse og Omdanning av Visse Aminosyrer
Aminosyrene i kroppen er essensielle både for oppbygging av proteiner og for regulering av viktige biokjemiske prosesser, inkludert fjerning av giftige forbindelser som ammonium. Glutamat er et godt eksempel på en aminosyre som spiller en viktig rolle i denne prosessen, spesielt på grunn av dens evne til å «fange» ammonium, som ellers kan være giftig i høye konsentrasjoner.
Reduktiv Aminering: Fanging av Ammonium
En viktig mekanisme for å håndtere ammonium i kroppen er reduktiv aminering. Denne prosessen skjer når α-ketoglutarat, et mellomprodukt i sitronsyresyklusen, kombineres med en aminogruppe (NH₄⁺) ved hjelp av enzymet glutamat dehydrogenase. Dette krever energi i form av NADPH (Nikotinamid-adenindinukleotidfosfat), som gir den nødvendige reduksjonsenergien.
I denne prosessen omdannes α-ketoglutarat til glutamat, og den giftige ammoniumionen bindes på en sikker måte, som forhindrer at ammonium bygger seg opp til giftige nivåer i kroppen.
Denne prosessen er en viktig del av kroppens strategi for å redusere toksisitet fra ammonium, og den kan reverseres i en prosess kalt oksidativ deaminering, som spiller en nøkkelrolle i nedbrytning av aminosyrer.
Amidering: Dannelse av Glutamin
Glutamat kan videre omdannes til glutamin i kroppen gjennom en prosess kalt amidering. Her festes en ekstra aminogruppe til glutamat ved hjelp av enzymet glutaminsyntetase, som bruker ATP som energikilde.
Glutamin er en tryggere form for transport av ammonium i kroppen. Det er spesielt viktig for vev som trenger å transportere nitrogen uten at det blir giftig, som for eksempel i nyrene og hjernen. Glutamin fungerer som en nitrogenbærer, og det er viktig for opprettholdelsen av metabolsk balanse.
Funksjonen til amidering: Amideringsprosessen er essensiell i kroppens håndtering av nitrogen, og den spiller en avgjørende rolle i å opprettholde kroppens metabolisme og i forebyggingen av ammoniakktoksisitet.
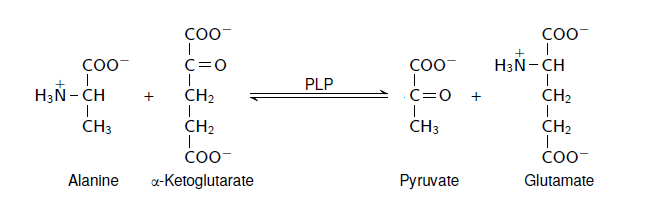
Transaminering: Overføring av Aminogrupper
En annen viktig prosess i aminosyremetabolismen er transaminering, hvor en aminogruppe overføres fra én aminosyre til en α-ketosyre. Dette er en reversibel prosess som gjør at kroppen kan danne nye aminosyrer etter behov.
Glutamat fungerer som en hovedgiver av aminogrupper i transamineringen, mens α-ketoglutarat ofte aksepterer aminogruppen for å danne glutamat.
Enzymer som er involvert i transamineringen inkluderer aminotransferaser, som ALAT (alanin-aminotransferase) og ASAT (aspartat-aminotransferase). Disse enzymene krever pyridoksalfosfat (vitamin B6) som kofaktor for å kunne utføre reaksjonene.
Eksempler på transaminering:
- ALAT katalyserer overføringen av en aminogruppe fra alanin til α-ketoglutarat, og dannelsen av pyruvat og glutamat. Forhøyede nivåer av ALAT i blodet kan indikere leverskade eller andre leverrelaterte sykdommer.
- ASAT overfører en aminogruppe fra aspartat til α-ketoglutarat, og danner oksaloacetat og glutamat. Forhøyede nivåer av ASAT kan indikere hjertesykdom, pankreatitt eller leversykdom.
Klinisk: Føllings Sykdom
Føllings Sykdom (Fenylketonuri, PKU)
Fenylketonuri, også kjent som Føllings sykdom, er en sjelden, medfødt metabolsk lidelse som påvirker kroppens evne til å bryte ned aminosyren fenylalanin. Denne sykdommen skyldes en mangel på enzymet fenylalanin-hydroksylase (PAH), som normalt omdanner fenylalanin til en annen aminosyre, tyrosin. Når dette enzymet mangler, akkumulerer fenylalanin i kroppen til potensielt giftige nivåer, noe som kan føre til alvorlige helseproblemer, spesielt i hjernen.
Årsak og genetikk
Føllings sykdom er forårsaket av mutasjoner i PAH-genet, som kodene for enzymet fenylalanin-hydroksylase. Denne genfeilen gjør at enzymet ikke kan utføre sin normale funksjon. PAH-mangelen fører til at fenylalanin ikke kan omdannes til tyrosin, som er viktig for flere fysiologiske prosesser i kroppen, inkludert produksjon av nevrotransmittere som dopamin og norepinefrin.
Fenylketonuri følger en autosomal recessiv arvegang, hvilket betyr at en person må arve en mutert kopi av PAH-genet fra begge foreldrene for å utvikle sykdommen. Hvis en person bare arver ett mutert gen, vil de være bærere av sykdommen, men de vil ikke vise symptomer.
Konsekvenser av Føllings sykdom
Når enzymet fenylalanin-hydroksylase ikke fungerer som det skal, fører det til en opphopning av fenylalanin i blodet og vevene. Dette skaper en rekke fysiologiske problemer, hovedsakelig relatert til nevrologisk utvikling.
- Opphopning av fenylalanin
- Fenylalanin brytes ikke ned og akkumulerer i blodet, noe som fører til høye nivåer i vev og organer. Dette kan føre til toksiske effekter, spesielt på hjernen.
- Omdanningsprodukter
- Fenylalanin som ikke kan omdannes til tyrosin, omdannes til biprodukter som skilles ut i urinen. Selv om dette gir en form for utskillelse, er det ikke nok til å hindre opphopningen i kroppen.
- Blokkering av andre aminosyrer
- Høye nivåer av fenylalanin kan blokkere transporten av andre essensielle aminosyrer over blod-hjerne-barrieren. Dette hindrer hjernen i å motta nødvendige byggesteiner for viktige molekyler, som nevrotransmittere. Dette fører til ytterligere nevrologiske problemer.
- Nevrologiske skader
- Den største konsekvensen av den ubehandlede sykdommen er dens virkning på hjernen. Fenylalanin virker toksisk på hjernen, og høye nivåer kan føre til:
- Hjerneskade
- Utviklingshemming, inkludert betydelig læringsvansker, atferdsproblemer og svekket kognitiv funksjon.
- Motoriske problemer: Langvarig fenylalanin-akkumulering kan føre til nedsatt motorikk og koordinasjon.
- Den største konsekvensen av den ubehandlede sykdommen er dens virkning på hjernen. Fenylalanin virker toksisk på hjernen, og høye nivåer kan føre til:
Diagnostisering og behandling
Føllings sykdom kan diagnostiseres tidlig gjennom nyfødtscreening for fenylalanin-nivåer i blodet. Tidlig påvisning og behandling er avgjørende for å unngå de alvorlige nevrologiske konsekvensene som kan følge med sykdommen.
Behandling består hovedsakelig i et fenylalaninfattig kosthold, som hindrer opphopning av fenylalanin i kroppen. Pasienter med PKU unngår matvarer som inneholder fenylalanin, som proteinrike matvarer som kjøtt, fisk, egg, melk, og soya. Spesialformulerte kosttilskudd og matvarer er tilgjengelige for å sikre tilstrekkelig ernæring uten overskudd av fenylalanin.
Enkelte pasienter kan også behandles med medisiner, som sapropterin, som kan hjelpe PAH-enzymet til å fungere mer effektivt i kroppen, og dermed redusere fenylalaninnivåene.
Prognose ved behandling
Ved tidlig diagnose og korrekt behandling kan personer med PKU leve et normalt liv uten nevrologiske problemer. Det er viktig å følge kostholdsrestriksjonene gjennom hele livet, ettersom høye fenylalaninnivåer kan skade hjernen selv i voksen alder. Med behandling kan de fleste med PKU føre et friskt liv og unngå de alvorlige konsekvensene som oppstår ved ubehandlet sykdom.
Læringspoeng
- Tidlig diagnose er avgjørende: PKU kan forhindres fra å forårsake alvorlige nevrologiske skader dersom sykdommen fanges opp tidlig gjennom nyfødtscreening.
- Kostholdsstyring er essensielt: Fenylalaninfattig kosthold er hovedbehandlingen, og det er viktig for pasienter å følge dette strengt gjennom hele livet for å unngå helsemessige problemer.
- Medisinsk behandling kan støtte kostholdet: Medisiner som sapropterin kan hjelpe til med å redusere fenylalaninnivåene hos noen pasienter, men kostholdet er fortsatt nøkkelen til å kontrollere sykdommen.
Føllings sykdom illustrerer hvordan genetiske sykdommer kan ha store konsekvenser for metabolske prosesser, og hvordan tidlig behandling og kostholdsstyring kan forhindre alvorlige helseskader.
📚 Anki-kort
Obs, tomt! Kommer etterhvert <3
📝 Eksamensoppgaver
Obs, tomt! Kommer etterhvert <3
👨⚕️ Klinisk case
Obs, tomt! Kommer etterhvert <3