Hva er hjertehypertrofi?
Hjertehypertrofi betyr at hjertemuskelen vokser i størrelse – nærmere bestemt at de enkelte hjertemuskelcellene (kardiomyocytter) øker i volum. Dette er kroppens naturlige måte å tilpasse seg økt belastning på. Når hjertet over tid må jobbe hardere, enten på grunn av fysiologiske krav eller patologiske forhold, vil det forsøke å styrke seg selv – akkurat som en vanlig muskel ville gjort.
Men ikke all vekst er god vekst.
Det finnes nemlig to former for hjertehypertrofi, og de har svært forskjellige konsekvenser:
- Fysiologisk hypertrofi oppstår som en sunn og reversibel tilpasning – for eksempel ved utholdenhetstrening eller under graviditet. Hjertet blir sterkere, større og mer effektivt, uten å miste funksjonen.
- Patologisk hypertrofi derimot, er en usunn og ofte progressiv prosess som skjer som følge av sykdom, for eksempel ved langvarig høyt blodtrykk eller etter et hjerteinfarkt. Her ser vi strukturelle forandringer, fibrose og redusert pumpeevne – og i verste fall utvikling av hjertesvikt.
💡 Hypertrofi betyr ikke alltid det samme. Samme ord – men vidt forskjellig virkning.
Å forstå forskjellen på disse to variantene er avgjørende i både fysiologi og klinikk. Det hjelper oss å vite når hjertet tilpasser seg normalt, og når det begynner å feile.
Fysiologisk hypertrofi – hjertets sunne tilpasning
Når hjertet blir sterkere av trening, er det ikke snakk om sykdom eller overbelastning, men om en sunn tilpasning – en fysiologisk hypertrofi. Hjertet tilpasser seg en økt belastning på en måte som gjør det mer effektivt. Det vokser, ja, men dette skjer kontrollert og uten at det oppstår skade, fibrose eller svekket funksjon.
Tvert imot: funksjonen forbedres. Hjertet slår kraftigere, fyller seg bedre, og tømmer seg mer effektivt – alt uten å miste sin elastisitet og evne til å slappe av mellom slagene.
Denne typen tilpasning skjer hos utholdenhetsutøvere som løpere, syklister og svømmere, men også hos gravide kvinner, hvor hjertet må jobbe hardere for å dekke både morens og fosterets behov for oksygen og næring. Det er altså en reversibel, gunstig tilstand, og det motsatte av det vi ser ved hjertesvikt eller patologisk belastning.
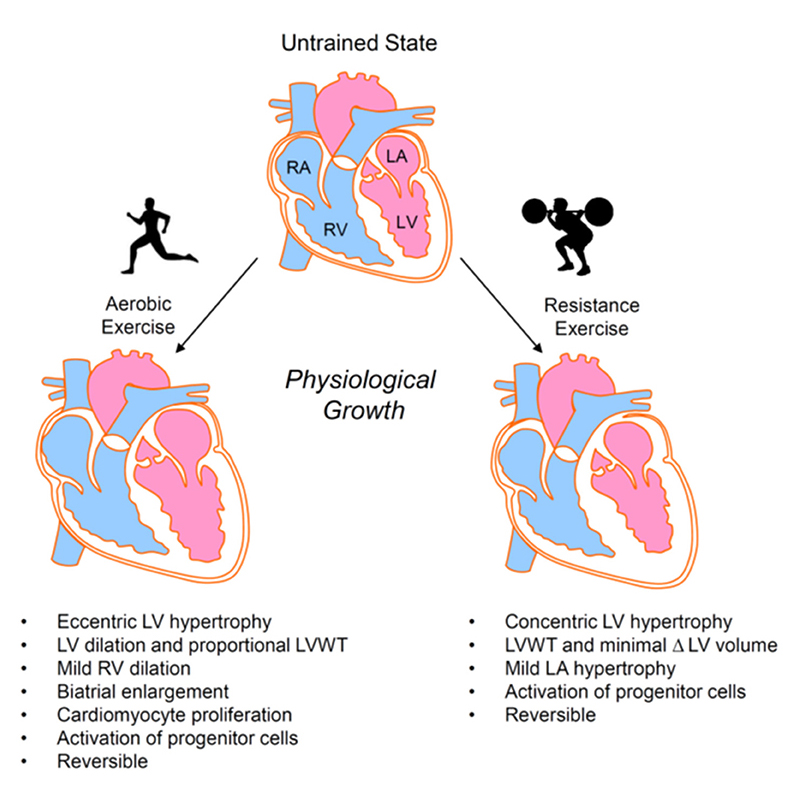
Ulike typer trening gir ulike hjertestrukturer
Måten hjertet tilpasser seg på, avhenger sterkt av typen belastning.
Ved utholdenhetstrening – som jogging, langrenn eller svømming – øker først og fremst volumet hjertet må håndtere. Hjertets kammer, særlig venstre ventrikkel, utvider seg, og myocyttene vokser i lengde.
Denne formen for utvidelse kalles eksentrisk hypertrofi, og gir et hjerte som fyller seg mer i diastolen, og derfor kan pumpe ut mer blod i hvert hjerteslag.
Styrketrening gir derimot en annen type belastning. Når man løfter tungt, øker blodtrykket dramatisk i korte øyeblikk, og dette gir hjertet høyere trykkmotstand – altså økt afterload.
For å kompensere, blir veggene i hjertets venstre ventrikkel tykkere, uten at selve kammeret nødvendigvis blir større.
Dette kalles konsentrisk hypertrofi, og gir kraftigere kontraksjoner – men ikke nødvendigvis økt slagvolum. Hjertemuskelcellene vokser da i bredden, ikke i lengden.
Hva skjer med slagvolumet?
Trente personer har som regel lavere hvilepuls, og det skyldes at hvert hjerteslag pumper ut mer blod enn hos utrente. Dette høyere slagvolumet skyldes både strukturelle og funksjonelle tilpasninger. Når hjertet fylles bedre, og samtidig trekker seg kraftigere sammen, blir det mer effektivt. Diastolen blir lengre – fordi pulsen er lavere – og dette gir hjertet mer tid til å fylle seg før neste slag. Samtidig har hjertemuskelen blitt mer følsom for kalsium og mer effektiv i kontraksjonen. Til sammen betyr dette at hjertet bruker færre slag på å gjøre den samme jobben.
Denne effektive pumpemekanismen kommer av både flere sarkomerer i serie (som gjør hjertet større) og bedre evne til å regulere ioneflyt, særlig kalsium og natrium, som styrer sammentrekning og avslapning.
Endringer i hjertemuskelcellene
Hjertemuskelcellene – myocyttene – vokser altså i lengde eller bredde, avhengig av belastningstypen.
Ved utholdenhetstrening ser man at cellene blir lengre(sarkomer legger seg i serie) og kan forkorte seg mer under kontraksjon.
Dette gir mer kraft per celle.
Myocyttene får også bedre evne til å håndtere kalsium, som er helt avgjørende for både sammentrekning og avslapning. Kalsium pumpes raskere tilbake i lagrene etter hvert slag, og cellen er raskt klar for neste kontraksjon.
Sammen gir dette et hjerte som slår kraftigere, roligere og mer utholdende – et hjerte i toppform, bokstavelig talt.
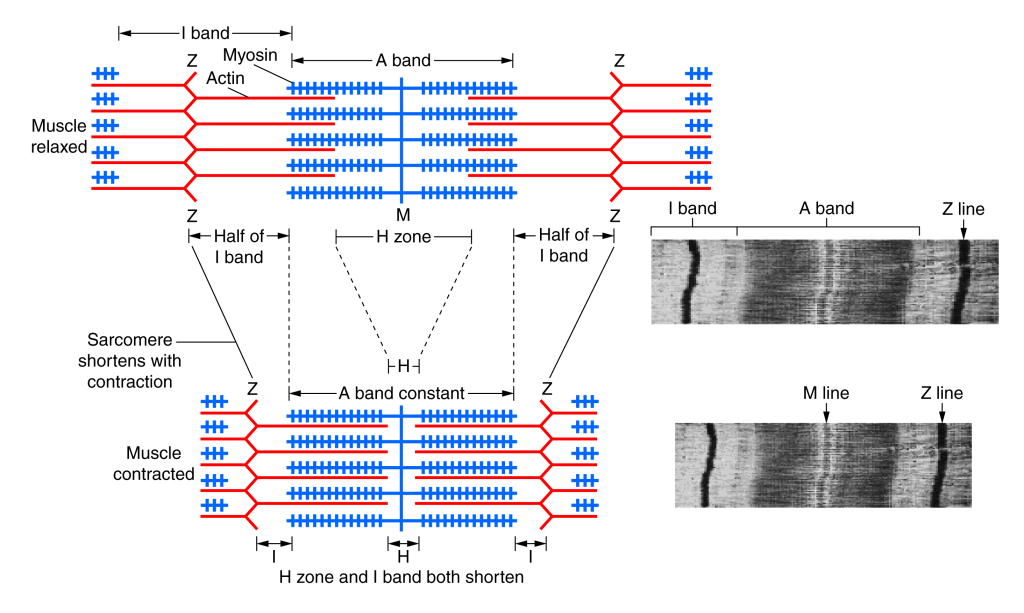
Ved styrketrening, der hjertet møter kortvarig, men kraftig trykkbelastning – som når man løfter vekter – skjer det en annen type vekst. I stedet for at myocyttene vokser i lengde, som ved utholdenhetstrening, vokser de i bredde.
Dette skjer ved at sarkomerer legges til i parallell, altså ved siden av hverandre, som ekstra lag i tykkelsen av muskelfibrene. Hjertemuskelcellene blir dermed tykkere, og veggene i særlig venstre ventrikkel øker i tykkelse.
Denne typen vekst – konsentrisk hypertrofi – gir hjertet evnen til å utvikle høyere trykk, fordi hver celle får flere kontraktile enheter side om side, som samtidig drar i hver sin aktintråd. Dette gir et kraftigere slag, men uten nødvendigvis å øke hvor mye blod hjertet kan holde – altså ingen økt fyllingskapasitet.
I motsetning til utholdenhetstilpasningen, hvor hjertet får større volum, gir styrketrening et kompakt og trykksterkt hjerte – godt egnet til å takle korte og intense belastninger.
Nå som vi forstår hva som skjer strukturelt og funksjonelt ved fysiologisk hypertrofi, er neste spørsmål: Hva er det som gir beskjed til hjertet om å vokse? Hvordan vet kroppen at belastningen er økt, og hvilke signalveier aktiveres for å bygge et større og sterkere hjerte?
Nå skal vi inn på de viktigste signalveiene.
Signalveier og molekylære mekanismer bak fysiologisk hypertrofi
Fysiologisk hypertrofi er en strukturert og nøye regulert prosess hvor hjertet tilpasser seg økt belastning – som ved trening eller graviditet – uten å utvikle fibrose eller nedsatt pumpefunksjon.
For at hjertemuskelen skal vokse på en sunn måte, må det skje endringer helt ned på cellenivå, der spesifikke signalveier styrer både genuttrykk og proteinsyntese.
IGF-1 og PI3K–Akt–mTOR: Kjerneveien bak fysiologisk hjertesvekst
Et sentralt signalmolekyl i denne prosessen er IGF-1 (insulin-like growth factor 1). Dette proteinet produseres både i leveren og lokalt i hjertemuskulaturen, og nivåene øker ved fysisk trening.
Når IGF-1 binder seg til sin reseptor på hjertemuskelcellene, IGF-1-reseptoren (IGF1R), aktiveres en signalvei som er avgjørende for å fremme fysiologisk vekst: PI3K–Akt–mTOR. Om du skal kunne en signalvei, så er det denne.
Trinnene i signalveien:
- PI3K (fosfatidylinositol-3-kinase) aktiveres og viderefører signalet inn i cellen.
- PI3K aktiverer Akt, et nøkkelprotein som har flere viktige funksjoner:
- Akt aktiverer mTOR (mammalian target of rapamycin), et regulatorisk protein som stimulerer proteinsyntese.
- Akt hemmer GSK-3β (glycogen synthase kinase 3 beta), et protein som normalt begrenser cellevekst. Når det hemmes, øker cellens evne til å bygge opp kontraktile proteiner.
mTOR aktiverer blant annet S6 kinase (S6K1) og 4E-BP1, som begge fremmer translasjon av vekstrelaterte gener.
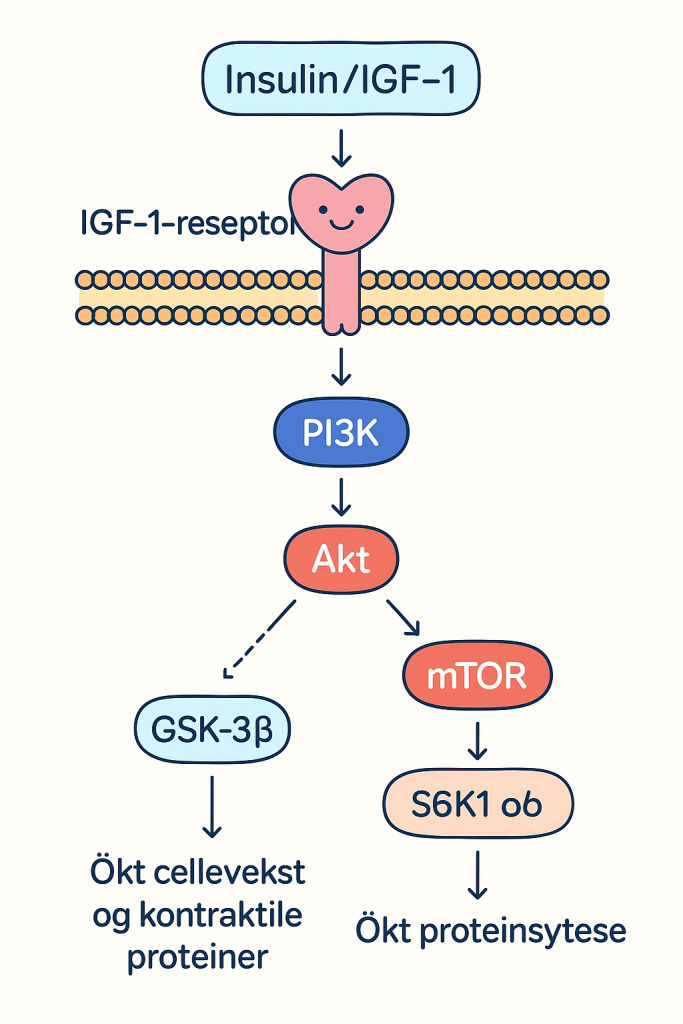
Resultatet er at hjertemuskelcellene øker i størrelse gjennom økt proteinsyntese og økt produksjon av kontraktile strukturer – uten tegn til patologisk aktivering eller celledød.
Supplerende signalveier
I tillegg til PI3K-Akt-mTOR-veien, finnes det flere supplerende mekanismer som bidrar til en sunn hypertrofisk respons:
- JAK/STAT-signalveien og PGC-1α regulerer mitokondriefunksjon og energimetabolisme, og gjør hjertemuskulaturen mer utholdende.
- Thyreoideahormoner og vekstfaktorer som VEGF og FGF fremmer differensiering og tilpasning til fysisk belastning.
Disse signalene arbeider parallelt med PI3K-Akt-mTOR for å sikre optimal vekst uten strukturell skade.
Hos trente personer ser man i tillegg lavere nivåer av signalstoffer som normalt er forbundet med patologisk hypertrofi. Blant disse finner vi angiotensin II og endotelin-1, som begge kan bidra til fibrose og negativ ombygging ved hjertesykdom. Reduksjonen i disse stoffene antyder at fysiologisk hypertrofi også innebærer en nedregulering av skadelige signaler.
Konsentrisk og eksentrisk hypertrofi
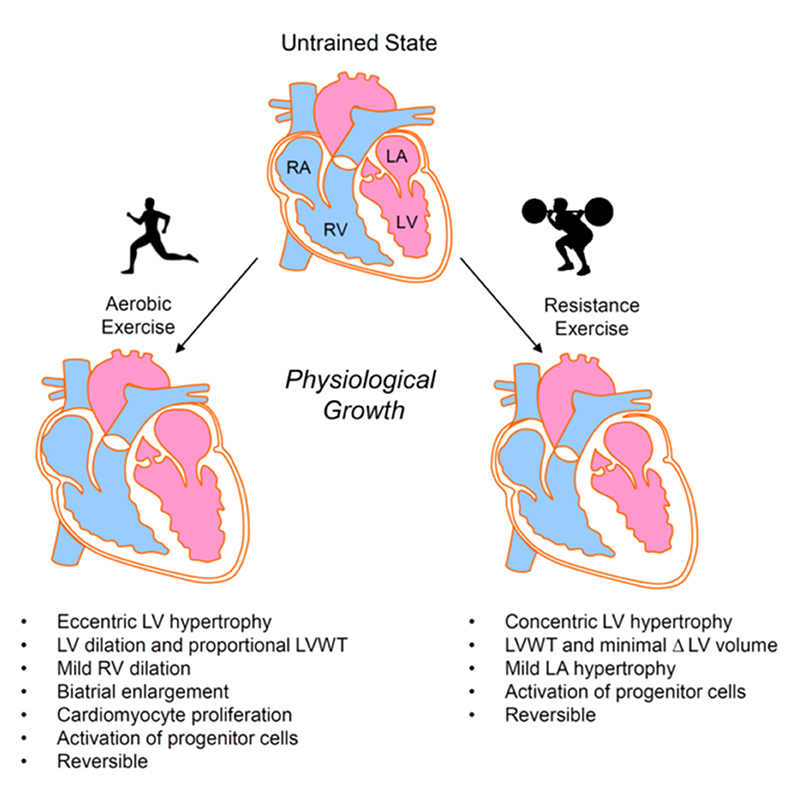
Hjertemuskelen er ikke bare et organ som jobber hardt – det er også en fantastisk tilpasningsdyktig muskel.
Når den møter vedvarende belastning, prøver den å tilpasse seg.
Men hvordan den tilpasser seg, avhenger helt av hva slags belastning det er snakk om. Det finnes to hovedtyper av hypertrofi: konsentrisk og eksentrisk. Og forskjellen handler både om hva som skjer med veggtykkelsen og med volumet inni hjertet.
Konsentrisk hypertrofi
Konsentrisk hypertrofi oppstår når hjertet stadig må jobbe mot økt trykk – for eksempel ved høyt blodtrykk (hypertensjon) eller ved aortastenose (forsnevret aortaklaff). Det betyr at hjertet må presse blodet ut mot mer motstand. Hvordan løser hjertemuskelen dette?
Jo, den øker veggtykkelsen, uten at volumet inni kammeret nødvendigvis øker. Det skjer ved at hjertemuskelcellene (kardiomyocyttene) legger til flere sarkomerer i parallell. Tenk deg at de vokser «bredere» – som en bodybuilder som trener på å løfte tungt. Hjertet får mer muskelmasse, men ikke nødvendigvis bedre funksjon.
Konsekvensen? Veggen blir tykk, men elastisiteten og fyllingsevnen kan svekkes. Med tiden kan det føre til diastolisk svikt – hjertet klarer ikke slappe godt nok av og fylle seg.
Eksentrisk hypertrofi
Eksentrisk hypertrofi oppstår når hjertet over tid må håndtere økt volum – som ved klaffeinsuffisiens eller etter et hjerteinfarkt der veggen svekkes og dilaterer. Her trenger hjertet å kunne romme og pumpe mer blod.
Hvordan responderer det? Det øker både volum og veggtykkelse, men først og fremst volum. Hjertemuskelcellene legger til flere sarkomerer i serie, altså lengde, slik at cellene strekker seg og ventrikkelen blir større.
Dette gir et hjerte som kan pumpe større mengder blod, men som også kan bli mer slapt og mindre effektivt hvis belastningen fortsetter. På sikt kan dette føre til dilatert hjertesvikt – hvor hjertet blir som en overfylt og utstrekt ballong som ikke klarer å trekke seg sammen ordentlig.
Sammenligning av konsentrisk og eksentrisk hypertrofi
| Egenskap | Konsentrisk hypertrofi | Eksentrisk hypertrofi |
|---|---|---|
| Årsak | Økt trykk (afterload) | Økt volum (preload) |
| Sarkomer-vekst | I parallell (bredde) | I serie (lengde) |
| Resultat | Tykkere vegg, lite endret volum | Økt volum, tynnere vegg (relativt) |
| Eksempel | Hypertensjon, aortastenose | Klaffeinsuffisiens, infarkt, utholdenhetstrening |
| Risiko | Diastolisk svikt | Systolisk svikt ved dilatasjon |
💡 Hovedpoeng: Konsentrisk og eksentrisk hypertrofi er to ulike strategier hjertet bruker for å takle økt belastning. Men bare én av dem – den vi ser ved aerob trening – er egentlig sunn og reversibel. Den andre er en varsellampe på at hjertet sliter med å tilpasse seg.
Patologisk hypertrofi og veien mot hjertesvikt
Når hjertet utsettes for langvarig og unormal belastning, som ved høyt blodtrykk, klaffesykdom eller etter et hjerteinfarkt, settes det i gang kompensasjonsmekanismer som i starten kan være nyttige. Hjertemuskelen forsøker å tilpasse seg ved å vokse – men denne veksten har en annen karakter enn den vi ser ved trening. Den blir gradvis skadelig, og kan over tid føre til hjertesvikt.
Myokardisk skade og remodellering
Et hjerteinfarkt er kanskje den mest dramatiske årsaken til patologisk remodellering. Når blodtilførselen til deler av hjertet kuttes av, dør myokardceller, og det oppstår en betennelsesreaksjon som setter i gang vevsombygging. Dette skjer i flere faser:
- Akutt fase (timer): Hjertemuskelceller nekrotiserer (dør), og området preges av inflammasjon.
- Infarktekspansjon (dager): Det skadede området blir større, og veggen i dette området svekkes og utvides.
- Global remodellering (uker–måneder): Hele hjertet begynner å endre form og funksjon – volumet øker, men kontraktiliteten svekkes.
Resultatet er et dilatert hjerte med dårlig pumpefunksjon. Det pumper mer blod inn, men klarer ikke tømme seg effektivt. Hjertets form endres fra en stram, konisk muskel til en mer avrundet, slapp ballong – et tydelig tegn på patologisk remodellering.
Overbelastning → Hypertrofi → Hjertesvikt
Dette forløpet gjelder ikke bare infarkt, men også ved langvarig trykk- eller volumbelastning. Når hjertet stadig må jobbe mot høyt trykk (som ved hypertensjon eller aortastenose), eller stadig må håndtere store blodvolumer (som ved klaffeinsuffisiens), svarer det med å bygge mer muskel. Men dette er ikke bærekraftig på lang sikt.
Kompenserende vs. maladaptiv hypertrofi
I begynnelsen kan denne hypertrofien være en god ting – veggtykkelsen øker for å normalisere veggspenningen (jfr. LaPlaces lov). Hjertemuskelen forsøker å opprettholde slagvolum. Men etter hvert som belastningen vedvarer, slår denne tilpasningen over i det motsatte:
- Muskelcellene vokser, men ikke koordinert
- Energiomsetningen svekkes
- Fibrose (arrvev) begynner å erstatte normalt vev
- Elektrisk ledning blir ujevn, og risiko for arytmier øker
- Kontraktiliteten faller – og vi får hjertesvikt
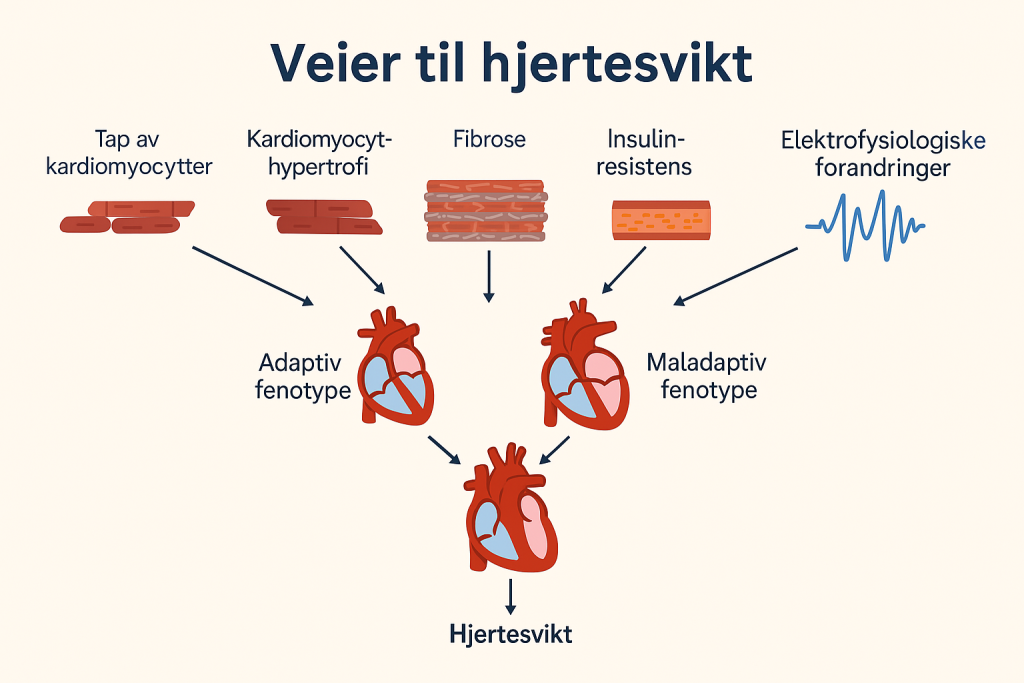
Patologisk vs. fysiologisk hypertrofi – hva er forskjellen?
| Egenskap | Fysiologisk hypertrofi | Patologisk hypertrofi |
|---|---|---|
| Årsak | Trening, graviditet | Hypertensjon, infarkt, klaffefeil |
| Genuttrykk | Normalt, adaptivt | «Fosterlignende», maladaptivt |
| Cellevekst | Koordinert, proporsjonal | Ukoordinert, ofte i bredde |
| Fibrose | Ingen | Ofte tilstede |
| Funksjon | Opprettholdt eller forbedret | Ofte svekket |
| Reversibilitet | Ja | Ofte nei, eller bare delvis |
💡 Hovedpoenget: I motsetning til den balanserte og reversible veksten ved fysiologisk hypertrofi, er den patologiske formen en skjev og ubalansert prosess som, hvis den ikke bremses, leder mot hjertesvikt og redusert livskvalitet.
Signalveier og reseptorer ved hypertrofi og hjertesvikt
Når hjertet utsettes for langvarig belastning – enten det skyldes høyt blodtrykk, volumoverbelastning, hjerteinfarkt eller andre former for stress – aktiveres et komplekst samspill av signalveier. Dette er ikke tilfeldige prosesser. Hjertemuskelcellene «hører» på signaler fra både nerver, hormoner og mekanisk stress, og svarer med endringer i vekst, funksjon og genuttrykk.
Noen av disse signalene er tilpasningsdyktige – som vi ser ved fysiologisk hypertrofi ved trening. Andre fører til ubalanse, arrvev og tap av funksjon – det vi kaller patologisk remodellering. Forskjellen ligger i hvilke reseptorer som aktiveres, og hvilke signalveier de setter i gang.
På overflaten av kardiomyocyttene finner vi mange forskjellige reseptorer, og de fungerer som små antenner. Når disse reseptorene fanger opp signaler fra for eksempel hormoner eller inflammatoriske stoffer, starter det en rekke intracellulære reaksjoner – såkalte signalveier – som til slutt påvirker hjertets struktur og funksjon.
De viktigste signalveiene i hjertet
1. G-proteinkoblede reseptorer (GPCR)
Disse reseptorene er sentrale i hjertets respons på nevrohormonell aktivering. Noradrenalin, adrenalin, angiotensin II og endotelin er eksempler på signalstoffer som binder seg til disse reseptorene.
- Når noradrenalin binder seg til en β₁-adrenerg reseptor, aktiveres en signalkaskade som øker produksjonen av cAMP og aktiverer PKA – noe som styrker kontraktilitet og korter ned relaksasjonstiden.
- α₁-reseptorer bruker en annen vei: De aktiverer fosfolipase C, som øker nivåene av IP₃ og DAG. Dette fører igjen til økt intracellulært kalsium og aktivering av PKC – et signal som bidrar til hypertrofi.
- AT₁-reseptoren for angiotensin II og ETA-reseptoren for endotelin fungerer på samme måte, men aktiverer hovedsakelig hypertrofiske og fibrotiske responser – og forbindes sterkt med hjertesvikt.
Dette kan du lese mer om på Signalsubstanser og nervrohormonell regulering.
2. Reseptor-tyrosin-kinaser (RTK)
Disse reseptorene aktiveres av vekstfaktorer, blant annet IGF-1 og bFGF.
Når de aktiveres, trigger de PI3K-Akt-mTOR-aksen – en signalvei som øker proteinsyntese, cellevekst og overlevelse. Denne typen signalering er sentral i fysiologisk hypertrofi, som ved trening, som vi har lest om tidligere.
3. Cytokinreseptorer og inflammasjon
Cytokiner som IL-6 og TNF-α binder til egne reseptorer og aktiverer JAK/STAT-signalveien.
Dette er særlig viktig ved hjertesvikt og patologisk hypertrofi.
IL-6 kan faktisk fremme hypertrofi direkte, mens TNF-α virker mer destruktivt – og forbindes med celledød og tap av kontraktilitet.
4. TGF-β og fibrose
Transforming Growth Factor Beta (TGF-β) er en annen aktør som spiller en sentral rolle i utvikling av fibrose – arrvev – i hjertet.
Når denne veien aktiveres, økes produksjonen av bindevevskomponenter, og hjertet blir stivere og mindre elastisk. Dette svekker pumpefunksjonen ytterligere.
5. Mekanisk stress og integriner
Når hjertet strekkes av høyt trykk eller store volumer, aktiveres også såkalte mekanosensoriske signalveier. Integriner i cellemembranen registrerer strekket og aktiverer Focal Adhesion Kinase (FAK), som igjen påvirker cellevekst og genuttrykk.
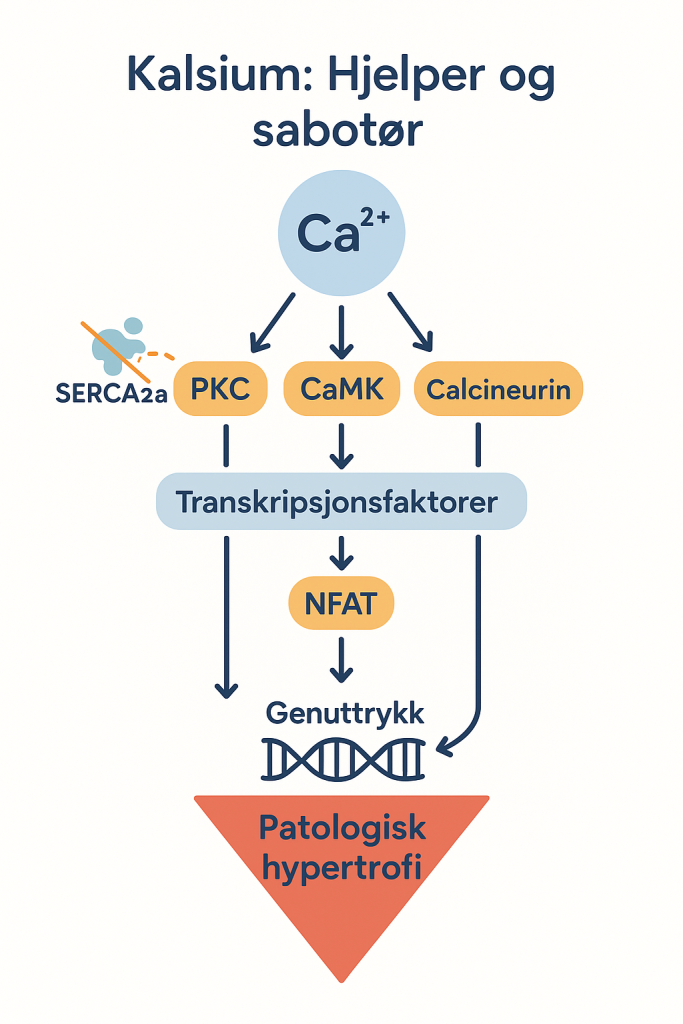
Kalsium: Destruktive signalveier
Kalsium er livsviktig for hver eneste hjertesammentrekning. Men når reguleringen av intracellulært kalsium svikter – som ved hjertesvikt – blir kalsium en del av problemet. En viktig feilmekanisme er svekket funksjon i SERCA2a-pumpen, som normalt frakter Ca²⁺ tilbake inn i sarkoplasmatisk retikulum etter hvert slag.
Når denne pumpen blir mindre effektiv, blir det liggende for mye kalsium i cytosol i diastolen. Resultatet er dårligere relaksasjon og energisløsing.
Samtidig aktiverer det høye kalsiumnivået en rekke signalveier:
- Calcineurin-NFAT-GATA4: Høyt Ca²⁺ aktiverer enzymet calcineurin, som igjen aktiverer transkripsjonsfaktoren NFAT.
Denne beveger seg inn i kjernen og slår på gener for vekst og hypertrofi – men på en måte som minner om fosterhjertet. Det gir strukturelle endringer som svekker hjertets funksjon. - PKC og CaM Kinase (CaMK): Begge disse veiene responderer på Ca²⁺ og aktiverer gener som endrer hjertemuskelcellens funksjon. Dette kan føre til både økt hypertrofi og tap av organisert struktur.
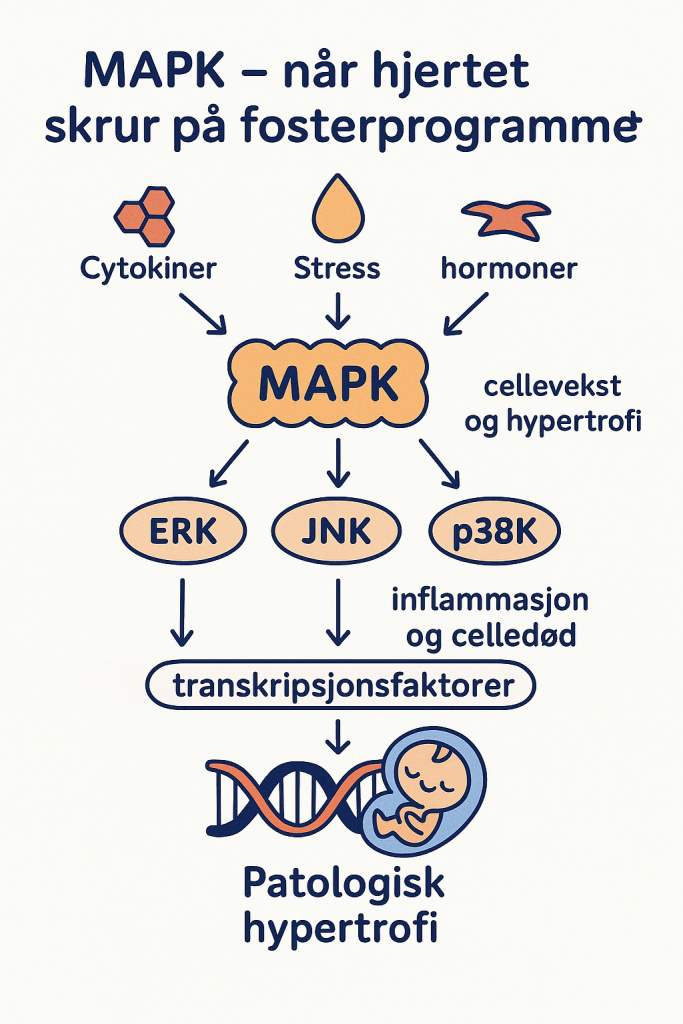
MAPK – Når hjertet skrur på «fosterprogrammet»
I møte med langvarig belastning, inflammasjon eller skade, kan hjertet aktivere signalveier som i utgangspunktet var viktige for fosterets utvikling. Den mest sentrale av disse er MAPK-aksen (Mitogen Activated Protein Kinase), og den spiller en nøkkelrolle i patologisk hypertrofi og hjertesvikt.
I fosterlivet er hjertet avhengig av raske cellevekstprogrammer for å kunne utvikles raskt og tilpasse seg et voksende miljø. I voksen alder, derimot, er disse programmene i stor grad slått av – fordi de ikke lenger trengs. Når hjertet skrur dem på igjen i voksen alder, er det sjelden et godt tegn.
Hva skjer når MAPK-veien aktiveres?
MAPK-aktivering fører til endret genuttrykk, der hjertet i økende grad:
- går over til en mindre effektiv energibruk
- produserer mer fibrotisk vev (bindevev)
- mister noe av sin evne til å trekke seg sammen effektivt
Det betyr at selv om hjertet vokser, blir det ikke sterkere – tvert imot. Det blir stivere, mer energikrevende, og mer sårbart for rytmeforstyrrelser og svikt.
De tre hovedgrenene i MAPK-systemet:
- ERK (Extracellular signal-Regulated Kinase)
ERK-aktivering er særlig koblet til cellevekst og kan bidra til hypertrofi, både fysiologisk og patologisk, avhengig av kontekst og signalstyrke. Ved kronisk stress skaper ERK mer ukontrollert cellevekst, som ikke nødvendigvis følges av tilsvarende funksjonell gevinst. - JNK (c-Jun N-terminal kinase)
JNK aktiveres spesielt ved oksidativt stress og inflammasjon, og er assosiert med celledød (apoptose) og remodellering. Når denne grenen aktiveres, mister hjertet både kontraktile enheter og elektrisk stabilitet – og blir gradvis svekket. - p38K (p38 kinase)
Også denne grenen responderer på stress og inflammasjon, og er særlig involvert i fibrose og tap av normale strukturer i hjertemuskelen. Aktivering av p38K er tett knyttet til proinflammatoriske cytokiner, som TNF-α og IL-6, og gir signaler om at hjertet skal ombygges – men dessverre i feil retning.
Klinisk betydning
MAPK-veien er altså et tveegget sverd: Den er essensiell for tilpasning i fosterlivet, men blir problematisk når den gjenaktiveres i voksen alder. I hjertesvikt ser man en oppregulering av flere av disse genene og veiene, og mange av endringene i hjertets struktur og funksjon kan forklares med økt aktivitet i MAPK-systemet.
💡 Oppsummert: MAPK-aktivering gir hjertet et slags “tilbakeskritt” i utvikling. Hjertet skrur på gamle vekstprogrammer som ikke passer i voksen alder – og resultatet er ofte fibrose, tap av kontraktilitet og en høyere risiko for hjertesvikt.
Viktig å få med seg: Reseptoravhengig vs. ikke-reseptoravhengig signalering
Når du får spørsmål om reseptormekanismer bak hjertets vekst, er det avgjørende å vite hvilke signalveier som faktisk starter i cellemembranen – altså via reseptorer – og hvilke som ikke gjør det.
Reseptoravhengige signalveier
Disse signalveiene er trigget av reseptorer som sitter i cellemembranen, og som gjenkjenner hormoner eller signalstoffer utenfra. Disse må kunne nevnes hvis spørsmålet handler om «reseptormekanisme»:
- IGF-1-reseptor → PI3K → Akt → mTOR
→ Fysiologisk hypertrofi
→ Reseptor: RTK (reseptor-tyrosin-kinase) - Angiotensin II → AT₁-reseptor (GPCR, Gq) → PLC → DAG/IP₃ → PKC → MAPK
→ Patologisk hypertrofi
→ Reseptor: G-proteinkoblet reseptor - Endotelin-1 → ETA-reseptor (GPCR) → samme kaskade som over
→ Patologisk hypertrofi
Ikke-reseptoravhengige signalveier
Noen signalveier er viktige for hypertrofi, men starter ikke i en reseptor i cellemembranen. Det betyr at de ikke kan regnes som «reseptormekanismer».
- ↑Ca²⁺ → Calcineurin → NFAT → GATA4
→ Patologisk hypertrofi
Ikke en reseptorstyrt vei – Ca²⁺ kommer fra innsiden og aktiverer et cytoplasmatisk enzym - CaMK → MEF2/GATA4
→ Patologisk hypertrofi
Igjen: ikke koblet til en spesifikk membranreseptor
📚 Eksempel fra eksamen
«Hva er den viktigste reseptormekanismen for hjertehypertrofien?»
→ Riktig svar må vise en membranreseptor
❌ Svar som starter med «økt intracellulært Ca²⁺…» er feil i denne sammenhengen
Husk:
| Signalvei | Starter i reseptor? | Hypertrofi-type |
|---|---|---|
| IGF-1 → PI3K → Akt → mTOR | ✅ Ja | Fysiologisk |
| Ang II / ET-1 → MAPK | ✅ Ja | Patologisk |
| Ca²⁺ → Calcineurin → NFAT | ❌ Nei | Patologisk |
| CaMK → MEF2 / GATA4 | ❌ Nei | Patologisk |
Oppsummert:
Hjertet formes og omformes av signaler fra omgivelsene. Det som avgjør om det tilpasser seg sunt eller utvikler sykdom, er hvilke reseptorer som aktiveres, og hvordan cellene responderer. Når sympatikus og angiotensin II dominerer over tid, settes maladaptive signalveier i gang som fører til fibrose, arytmier og svikt. Ved å forstå disse veiene, åpner vi også døren for målrettet behandling.
📚 Anki-kort
Obs, tomt! Kommer etterhvert <3
📝 Eksamensoppgaver
👨⚕️ Klinisk case
Obs, tomt! Kommer etterhvert <3